Single-nucleus RNA Sequencing Misses Activation of Human Microglia
Quick Links
In the presence of amyloid plaques, microglia in mouse and human brains react differently—or do they? In the September 29 Cell Reports, researchers led by Mark Fiers and Bart de Strooper at KU Leuven, Belgium, cast doubt on previous findings that human microglia do not activate the same genes in response to amyloidosis as do their mouse counterparts.
- Studies of AD brain use single-nuclei RNA-Seq to analyze microglial gene expression.
- But transcripts from activation genes are scarce in frozen microglial nuclei.
- This may cause snRNA-Seq to misread the microglial activation state.
Those previous expression studies were based on analyzing nuclei isolated from postmortem brain sections. Now, Fiers and colleagues report that transcripts of many microglial activation genes are relatively scarce in frozen microglial nuclei, but concentrated in the cytoplasm. Thus, those postmortem tissue studies easily could have missed an uptick in the expression level of these genes. The discrepancy might have masked microglial activation, the authors suggest. “At the moment, we don’t know what microglial activation in human brain looks like,” Fiers told Alzforum.
Other researchers agreed that this technical limitation could be a factor in the apparent difference between mouse and human microglial states. “This is an important study and makes a valid point,” said Oleg Butovsky at Brigham and Women’s Hospital, Boston. Likewise, Florent Ginhoux at Singapore’s Agency for Science, Technology and Research said the community should be aware of the limitations of existing methods. “[The data] stress the need to use complementary technologies, as well as the crucial need to validate as much as possible any conclusions drawn from such high-dimensional approaches,” he wrote to Alzforum (full comment below).
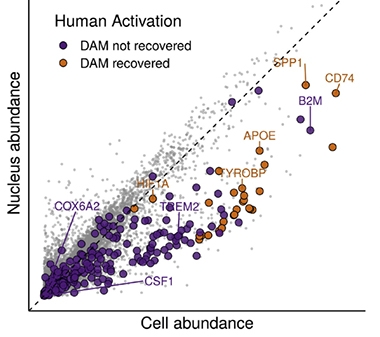
Nuclear Scarcity. Because disease-associated microglial (DAM) transcripts are in short supply in the nucleus, only the most abundant transcripts (gold) can be detected in nuclei from activated human microglia, while less-abundant (purple) ones are missed. [Courtesy of Thrupp et al., Cell Reports.]
Numerous studies have documented characteristic expression changes in mouse microglia around plaques. This profile has been variously labeled as disease-associated microglia (DAM), microglial neurodegenerative phenotype (MGnD), or amyloid-response microglia (ARM) (Jun 2017 news; Sep 2017 news; Apr 2019 conference news). Alas, in microglial nuclei isolated from postmortem AD brains, only a small subset of DAM genes are activated, and the exact ones vary from study to study (May 2019 news; Nov 2019 news; Jan 2020 news).
In unpublished work on AD brain, Fiers and colleagues likewise found few activated DAM genes. Curiously, however, human microglia grafted into a mouse model of amyloidosis switched on a higher number of these genes (Apr 2019 conference news; Mancuso et al., 2019). Was this activation due to the new environment of the mouse brain, or was it there all along in the human brain but somehow overlooked? Tissue preparation varies in studies of mouse and human microglia. The former are typically isolated from fresh brains using fluorescence-activated cell sorting, allowing researchers to analyze whole cells by single-cell RNA sequencing. Human microglia typically come from frozen postmortem sections, meaning the best option for bulk studies is to isolate nuclei for single-nuclei RNA-Seq.
To find out if the method made a difference, first author Nicola Thrupp directly compared single-cell and single-nuclei RNA-Seq of microglia obtained from temporal cortex biopsies taken from four people during epilepsy surgery. The patients were otherwise healthy and did not have Alzheimer’s disease. The authors isolated 14,823 whole cells for single-cell RNA-Seq, and 3,940 microglial nuclei from frozen sections for single-nuclei RNA-Seq.
Overall, the expression pattern for more than 98 percent of genes was similar in both the cell and nuclei sets. However, about 1 percent, or 246 transcripts, were scarce in nuclei. This gene set was consistent across all four brains examined. In addition, analysis of eight publicly available datasets of single-cell or single-nuclei RNA-Seq turned up the same set of nuclear-depleted transcripts.
These DAM Genes
Crucially, many of these nuclear-depleted transcripts encoded proteins involved in microglial activation. These included APOE, SPP1, CST3, FTL, PLD3, B2M, and CD74. If transcripts of these DAM genes are scarce in the nucleus, then would it be harder to detect microglial activation in nuclei from frozen AD brain sections? In support of this reasoning, a previous snRNA-Seq study of human microglia from AD brains had reported finding a subset of these genes elevated around plaques, but did not detect activation of the full set of DAM genes (Mathys et al., 2019). Tellingly, Thrupp and colleagues found that the transcripts detected in that study tended to be those with the highest overall expression level, while DAM transcripts with lower expression, such as TREM2, did not show up (see graph above).
This makes sense, Fiers said, because high-expressing genes might produce enough transcripts for single-nuclei RNA-Seq to reveal a boost in activation, even if those transcripts are relatively depleted in nuclei. Low-expressing genes, on the other hand, provide less statistical power to detect differential expression. “We think this is part of the reason why a number of studies have had a hard time seeing the full extent of microglial activation in the human AD brain,” Fiers said.
If so, how can researchers better detect DAM gene expression? Butovsky said that single-nuclei RNA-Seq remains the most viable option for expression studies of human brain. He believes isolating larger numbers of nuclei would boost statistical power, and speculated that if the authors had isolated as many microglial nuclei for analysis as they did whole cells, they might have seen fewer discrepancies between the expression profiles.
Fiers agrees that more nuclei and deeper RNA sequencing might help. He sees an alternative in spatial transcriptomics, which combines transcriptome analysis with in situ hybridization to reveal expression changes in individual cells in tissue slices. De Strooper’s lab has used this method to examine gene-expression changes in multiple cell types around amyloid plaques (Aug 2019 news; Chen et al., 2020).
Marco Colonna and Yingyue Zhou at Washington University in St. Louis agreed that technical limitations of single-nuclei RNA-Seq could cause false-negative results. “Validation of the differences identified by single-nuclei RNA-Seq at the protein level, such as by immunohistochemistry, becomes important,” they wrote to Alzforum (full comment below).
That said, they noted that Fiers and colleagues used only non-AD brain tissue for their study. In Alzheimer’s, the boost in DAM gene expression in activated microglia may make it easier to detect these transcripts, Colonna and Zhou suggested. For his part, Fiers wonders if amyloidosis might affect the distribution of DAM transcripts between nucleus and cytoplasm. He plans to compare the expression profiles of activated and homeostatic microglia using transgenic and chimeric mouse models.—Madolyn Bowman Rogers
References
News Citations
- Hot DAM: Specific Microglia Engulf Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
- Single-Cell Expression Atlas Charts Changes in Alzheimer’s Entorhinal Cortex
- Human and Mouse Microglia React Differently to Amyloid
- Chimeric Mice: Can They Model Human Microglial Responses?
- Spatial Transcriptomics Uncovers Coordinated Cell Responses to Amyloid
Paper Citations
- Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, Arranz-Mendiguren A, Sala Frigerio C, Claes C, Serneels L, Theys T, Perry VH, Verfaillie C, Fiers M, De Strooper B. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat Neurosci. 2019 Dec;22(12):2111-2116. Epub 2019 Oct 28 PubMed.
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019 Jun;570(7761):332-337. Epub 2019 May 1 PubMed.
- Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Laláková J, Kühnemund M, Voytyuk I, Wolfs L, Mancuso R, Salta E, Balusu S, Snellinx A, Munck S, Jurek A, Fernandez Navarro J, Saido TC, Huitinga I, Lundeberg J, Fiers M, De Strooper B. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer's Disease. Cell. 2020 Aug 20;182(4):976-991.e19. Epub 2020 Jul 22 PubMed.
Further Reading
Primary Papers
- Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, de Strooper B, Fiers M. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020 Sep 29;32(13):108189. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University School of Medicine
Washington University in St. Louis
Thrupp et al. propose that snRNA-Seq technology fails to recapitulate microglia activation states in humans because activation genes are underrepresented in non-diseased human brains by snRNA-Seq. The paper reveals a difference in sensitivity between scRNA-Seq and snRNA-Seq. This is an important observation that highlights a limitation of snRNA-Seq. However, as the authors state in the discussion, they mainly looked at non-diseased brains. If upregulation of activation genes can be detected in Alzheimer’s disease versus control by snRNA-Seq, even though the gene abundance is lower compared to scRNA-Seq, then snRNA-Seq technology should still be able to identify differentially expressed genes. In fact, some of the genes identified in this study as depleted in nucleus, such as APOE and TREM2, were identified as upregulated in AD microglia by snRNA-Seq in Zhou et al., 2020, and Mathys et al., 2019.
The authors propose that failure to identify DAM counterparts in human AD in previous studies was due to intrinsic drawbacks of snRNA-Seq. In agreement with this, we do not exclude the possibility that technical limitations caused by snRNA-Seq lead to false-negative results. In this context, validation of the differences identified by snRNA-Seq at protein level, such as by IHC, becomes important. It is also wise to incorporate results from various methodologies, for example, spatial transcriptomics as the authors indicated, when making conclusions.
To understand human diseases, it is important to identify changes between diseased and non-diseased tissues, hoping that these changes may provide new clues to disease mechanisms. Although scRNA-Seq may be more sensitive, it is not feasible in many cases, such as frozen autopsy specimens. Thus, although limited, snRNA-Seq remains a useful tool for detection of microglia activation.
References:
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M. Author Correction: Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med. 2020 Jun;26(6):981. PubMed.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Author Correction: Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019 Jul;571(7763):E1. PubMed.
Gustave Roussy Institute
This study shows that a small population of genes implicated in microglial activation, such as APOE, CST3, SPP1, and CD74, is depleted in “nuclei” versus “whole cell” single-cell RNA sequencing technologies. Importantly, these genes are part of the disease-associated microglia (DAM) program discovered by the laboratory of Ido Amit.
Although it is still unclear what the underlying reasons are for such differences, which the authors attribute to technical limitations inherent to snRNA-Seq, these findings underline that each technology on its own has clear advantages but also limitations. These are important to recognize, and openly discuss, in the community.
This important study also stresses the need to use complementary technologies, as well as the crucial need to validate as much as possible any conclusions drawn from such high-dimensional approaches.
Children's Hospital
Broad Institute of MIT and Harvard
Thrupp et al. compared single-nucleus (snRNA-Seq) and single-cell RNA-seq (scRNA-Seq) datasets generated from the temporal lobes of four human donors, and argue that the identification of microglial activation states—which are clearly implicated in AD pathogenesis—is impossible from single-nucleus data. The advent of high-throughput strategies for gene expression profiling of individual cells has opened up opportunities to characterize the cell states associated with Alzheimer’s disease. The use of single nuclei—which survive the freeze-thaw process intact—is potentially enormously enabling for these kinds of hypothesis-generating studies.
This study used early technical protocols for performing snRNA-Seq on human tissue, and the pace of methodological improvement over the past several years has been swift. Experimental innovations, led by participants in the Brain Initiative Cell Census Network (BICCN) consortium, in the isolation of individual nuclei for snRNA-Seq, combined with the use of more modern commercially available kits, has increased the median gene detection in brain nuclei more than threefold (Yao et al., 2020).
Computational strategies for integrative analysis that flexibly model and correct for systematic biases across datasets and commercial platforms (Welch et al., 2019; Stuart et al., 2019; Fleming et al., 2019) have made it considerably easier to appreciate glial activation states. For example, in our recent snRNA-Seq analysis of seven postmortem samples, we were able to identify a distinct microglial state associated with cerebral amyloid angiopathy, marked by upregulation of genes such as APOE and CST3 (Welch et al., 2019).
We remain optimistic that, with these recent methodological improvements, the field is well-positioned to utilize these new technologies to characterize microglial states within human tissue samples. Given the rapid pace of progress in this exciting area, open sharing of new analytical tools, methods, and data will be critical for the field to wage a concerted effort to decipher the complexity of the cellular mechanisms driving Alzheimer’s disease.
References:
Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell. 2019 Jun 13;177(7):1873-1887.e17. Epub 2019 Jun 6 PubMed.
Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019 Jun 13;177(7):1888-1902.e21. Epub 2019 Jun 6 PubMed.
Hong Kong University of Science & Technology
Characterizing the transcriptomic changes of AD patient brains at the single-cell level enables the identification of cell-type-specific dysregulated pathways in AD. While single-nucleus RNA-sequencing (snRNA-Seq) allows examining the transcriptomic change in frozen human brain tissues, this recent study raised concerns on whether snRNA-Seq can provide representative transcriptome profile for microglia. Thrupp et al. suggested that snRNA-Seq is suboptimal to study microglial activation, as some of the microglial activation signature genes (i.e. APOE, SPP1) are depleted in nuclei. The authors found that the transcripts of 246 genes (~1.1 percent of detectable genes) are more abundantly detected using single-cell RNA-Seq (scRNA-Seq) when compared to that of snRNA-Seq. Interestingly, many of these genes are the signature genes of disease-associated microglia, a specific subpopulation of microglia that associated with neurodegeneration. Thus, the authors raised a concern on the use of snRNA-Seq in detecting microglia activation in AD (Grubman et al., 2019; Mathys et al., 2019; Zhou et al., 2020).
While snRNA-Seq could potentially reduce the sensitivity of examining the microglial state transition, our recent study on the snRNA-Seq analysis of AD patient brains was able to show microglial activation in AD (Lau et al., 2020). Specifically, we found that microglia have increased expression of APOE, HLA-DQB1 and PTPRG in AD, in line with previous AD snRNA-Seq studies (Mathys et al., 2019). Although the transcript level of disease-associated microglia signature genes decreases in snRNA-Seq, these genes remain within the top 500 abundant genes in microglial nuclei. Thus, the transcriptome profiling by snRNA-Seq may be sufficient to illustrate the transition of microglial state in AD condition.
Indeed, our unpublished snRNA-Seq data can also demonstrate the microglial state transition in response to IL-33 (i.e., the induction of IL-33-responsive microglia) in an Aβ deposition mouse model, similar to what we have observed in our recent scRNA-Seq study (Lau et al., 2020). This shows that while snRNA-Seq is unable to capture the complete transcriptome signature of distinct activated microglia, we can still characterize the activation state of microglia by some representative signature genes in human brains. With further optimization of snRNA-Seq method and analysis, we are optimistic that we can study microglial state transition in AD using snRNA-Seq.
References:
Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJ, Petretto E, Polo JM. A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019 Dec;22(12):2087-2097. PubMed.
Lau SF, Cao H, Fu AK, Ip NY. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer's disease. Proc Natl Acad Sci U S A. 2020 Oct 13;117(41):25800-25809. Epub 2020 Sep 28 PubMed.
Lau SF, Chen C, Fu WY, Qu JY, Cheung TH, Fu AK, Ip NY. IL-33-PU.1 Transcriptome Reprogramming Drives Functional State Transition and Clearance Activity of Microglia in Alzheimer's Disease. Cell Rep. 2020 Apr 21;31(3):107530. PubMed.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH. Author Correction: Single-cell transcriptomic analysis of Alzheimer's disease. Nature. 2019 Jul;571(7763):E1. PubMed.
Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M. Author Correction: Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med. 2020 Jun;26(6):981. PubMed.
Make a Comment
To make a comment you must login or register.