Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
Quick Links
Scientists observe a mix of abnormally silent and overactive neurons in Alzheimer's disease brains, especially in the cortex. However, it is unclear which comes first—hyper- or hypoactivity—and why that might be. In a study reported in the May 16 Proceedings of the National Academy of Sciences USA, researchers led by Arthur Konnerth, University of Munich, Germany, removed the top layer of mouse brain to examine hippocampal activity in living transgenic mouse models of AD with two-photon microscopy. They report that the hippocampus revs up first before it quiets down, and place the blame for that altered activity on soluble Aβ oligomers.
"These data provide some of the strongest support to date for the hypothesis that Aβ species—likely soluble forms of Aβ—interfere with synaptic function in vivo and are associated with a hyperactive circuit within the hippocampus," Brad Dickerson, Massachusetts General Hospital, Charlestown, told Alzforum in an e-mail. Dickerson was not involved with the study.
A dampening of neural activity throughout the brain has long been recognized in AD (for a review, see Prvulovic et al., 2005). At the same time, more recent evidence, some of it from Konnerth's lab, suggests that neurons become overactive as well (see ARF related news story), especially near cortical plaques (see ARF related news story). In people, functional magnetic resonance imaging suggests that hippocampal hyperactivity puts people at risk for AD, according to Dickerson and colleagues (see ARF related news story). In addition, APP transgenic mice have silent seizures that may result from overexcitation and compensatory inhibition in entorhinal-cortical circuits (see ARF related news story on Palop et al., 2007). Could all this mean that neuronal hyperactivity comes first, and is an Aβ species to blame?
To investigate the question at the cellular level, first author Marc Aurel Busche and colleagues imaged the neural activity of living APP23xPS45 mice (see Busche et al., 2008), which form amyloid plaques at about three months of age. The scientists used a relatively new technique to observe the hippocampus, a brain area that is hit early on by AD pathology. They exposed the structure for two-photon microscopy by suctioning away a small piece of the overlying cortex in live mice held in place on the microscope. Then, focusing the microscope on hippocampal neurons, they could detect transient spikes in intracellular calcium, an indirect measure of neuronal firing. "We can now identify with single-cell resolution the location of the hyperactivity," said Christine Grienberger, who was not an author on this paper but is a member of Konnerth's lab and is involved with this work.
Older, six- to seven-month-old transgenic mice—in which many plaques had already developed—had more hypo- and hyperactive neurons than did wild-type mice (see image below). But in young, one- or two-month-old amyloid-free transgenic mice, neurons were mostly overactive compared to controls. There was even a trend toward fewer silent cells. This suggested an overall boost in hyperactivity that precedes plaque formation.
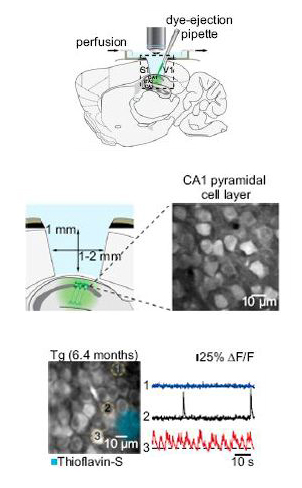
In-vivo two-photon microscopy of the mouse hippocampus
Removing a small part of the mouse cortex to expose the hippocampus (top) Konnerth and colleagues used two-photon microscopy to image neurons in layer CA1 of this brain region (middle). Measuring calcium transients (bottom), they found more hypoactive (cell No. 1, blue trace) and hyperactive (cell No. 3 and red trace) neurons than normal (Cell No. 2, black trace) in animals that had developed thioflavin S-positive amyloid deposits. Images courtesy of Arthur Konnerth. Copyright: National Academy of Sciences
If there were no plaques, what caused the overexcitation? To investigate, the researchers treated the mice with the γ-secretase inhibitor LY-411565. After one dose, soluble and insoluble hippocampal Aβ40 and Aβ42 fell and reached a low point about five hours after treatment; at the same time, the action potentials of CA1 neurons shrank almost to control levels. In contrast, the inhibitor didn't change action potential levels in wild-type mice. These results implicate soluble Aβ in the transgenic mouse's hyperactivity, the authors suggest.
Which Aβ species could be at fault? To see if a toxic Aβ species could directly change neural activity, the researchers bathed CA1 neurons of wild-type mice in synthetic Aβ40 dimers (see ARF related news story). Activity in the CA1 region of the hippocampus immediately shot up. Those results provide a causal link between soluble Aβ and hyperactivity, the authors wrote. "The experiment suggests the dimers could be the key species, but it doesn't exclude that other species could be involved," said Grienberger. Toxic Aβ42 dimers have been found in the human brain (see ARF related news story). It is unclear how soluble Aβ caused the heightened hippocampal activity, but one theory is that it boosts synaptic glutamate in the hippocampus, the authors wrote (see also ARF related news story). It might also contribute to a domino effect, suggested Dickerson. "It would be interesting to investigate whether the hyperactivation further promotes Aβ release and possibly fibrillar accumulation in a vicious cycle," he said.
Interestingly, in one- to two-month-old mice, cortical cells in the transgenic mice were about as active as in controls, implying that this pathology hits the hippocampus first. That result jibes with another recent paper in which Grienberger and colleagues demonstrated that plaques, not soluble Aβ, disrupt tuning of neurons in the visual cortex (see Grienberger et al., 2012). "It seems the cortex and hippocampus are very different in this respect," said Grienberger. When only soluble Aβ was present in the cortex, her group saw no changes in activity. The cortex may be better able to compensate for soluble Aβ, or perhaps the hippocampal cells are just more vulnerable, but the question will need further study, she said.
Hippocampal hyperactivity may be a therapeutic target, some recent research suggests. For instance, one study reported that reducing activity in hippocampal neurons with an epilepsy drug improves function on a memory task for people with aMCI (see ARF related news story). In addition, boosting inhibitory neurons improved network function and cognition in transgenic mice (see ARF related news story). The modified in-vivo two-photon microscopy used in the present study would allow researchers to look at single cell effects of such treatments, said Grienberger.—Gwyneth Dickey Zakaib
References
News Citations
- Hyperactive Neurons and Amyloid, Side by Side
- More Calcium News: Plaques Cause Dendrite Damage via Ion Overload
- Research Brief: Hippocampal Hyperactivity Tied to Early MCI Atrophy
- Do "Silent" Seizures Cause Network Dysfunction in AD?
- Aβ Neurotoxicity—Is it the Dimer? No, and Yes
- Patient Aβ Dimers Sufficient for Tau, Neuritic Changes
- Neuronal Glutamate Fuels Aβ-induced LTD
- Epilepsy Drug Calms the Hippocampus, Aids Memory
- Needs Salt: Reduced Sodium Channel Linked to Seizures in AD Model
Paper Citations
- Prvulovic D, Van de Ven V, Sack AT, Maurer K, Linden DE. Functional activation imaging in aging and dementia. Psychiatry Res. 2005 Nov 30;140(2):97-113. PubMed.
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007 Sep 6;55(5):697-711. PubMed.
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
- Grienberger C, Rochefort NL, Adelsberger H, Henning HA, Hill DN, Reichwald J, Staufenbiel M, Konnerth A. Staged decline of neuronal function in vivo in an animal model of Alzheimer's disease. Nat Commun. 2012;3:774. PubMed.
Further Reading
Papers
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012 May 10;74(3):467-74. PubMed.
- Rochefort NL, Jia H, Konnerth A. Calcium imaging in the living brain: prospects for molecular medicine. Trends Mol Med. 2008 Sep;14(9):389-99. PubMed.
- Mizrahi A, Crowley JC, Shtoyerman E, Katz LC. High-resolution in vivo imaging of hippocampal dendrites and spines. J Neurosci. 2004 Mar 31;24(13):3147-51. PubMed.
- Dombeck DA, Harvey CD, Tian L, Looger LL, Tank DW. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci. 2010 Nov;13(11):1433-40. PubMed.
News
- Chicago: AD and Epilepsy—Joined at the Synapse?
- Neuronal Glutamate Fuels Aβ-induced LTD
- Hyperactive Neurons and Amyloid, Side by Side
- Do "Silent" Seizures Cause Network Dysfunction in AD?
- More Calcium News: Plaques Cause Dendrite Damage via Ion Overload
- Chicago: AD and Epilepsy—Lessons from the Clinic, Animals
- New γ-Secretase Modulators Reduce Aβ42, Avoid Notch
- Do Overactive Brain Networks Broadcast Alzheimer’s Pathology?
- Research Brief: Hippocampal Hyperactivity Tied to Early MCI Atrophy
- Aβ Neurotoxicity—Is it the Dimer? No, and Yes
- Patient Aβ Dimers Sufficient for Tau, Neuritic Changes
- Epilepsy Drug Calms the Hippocampus, Aids Memory
- Needs Salt: Reduced Sodium Channel Linked to Seizures in AD Model
Primary Papers
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8740-5. Epub 2012 May 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Massachusetts General Hospital
These data provide some of the strongest support to date of the hypothesis that Aβ species—likely soluble forms of Aβ— interfere with synaptic function in vivo and are associated with a hyperactive circuit within the hippocampus. Further investigations of this animal model will likely provide important additional information about the mechanisms of aberrant physiology in the context of pre-plaque Aβ-related hippocampal dysfunction. It would be particularly interesting to investigate whether the hyperactivation further promotes Aβ release and possibly fibrillar accumulation in a vicious cycle. In addition, these findings further support the potential value of functional MRI markers of hippocampal hyperactivation in living humans with MCI as indicators of circuit dysfunction, and suggest that hippocampal hyperactivation should be investigated as a possible early marker of therapeutic response in clinical trials in which Aβ-modifying drugs are given to humans.
View all comments by Brad DickersonUniversity of Minnesota
In the past weeks, two studies using APPxPS1 transgenic animals describe abnormal calcium homeostasis as a potential early event in asymptomatic pre-plaque mice. Despite remarkable technical skills displayed by both teams, there studies might suffer from the same experimental confound.
The first report, by Arthur Konnerth’s group (Busche et al., 2012), is a follow-up study of previous work from the same group (Busche et al., 2008), which suggested the presence of clusters of hyperactive neurons near amyloid plaques in the bigenic APP23xPS45 mouse model. Using the same APP transgenic mice, this new article documents an impressive use of two-photon microscopy to investigate potential dysregulation of calcium signaling in hippocampal neurons in vivo. The authors report that, not only does apparent elevation of calcium signaling occur around plaques, but also that it takes place in younger, pre-plaque animals. Following the demonstration that spontaneous Ca2+ transients correspond to neuronal activity, Konnerth’s group then acutely applied the γ-secretase inhibitor LY-411575 to test whether Aβ was responsible for the observed effect. One single dose of the pharmacological agent reduced Aβ production and partly abolished neuronal hyperactivity in hippocampal neurons. The authors next applied synthetic Aβ dimers (AβS26C) at 100 nM for 30 seconds to hippocampal neurons in non-transgenic mice and observed a marked increase in Ca2+ transients. Busche and coworkers then concluded that soluble Aβ were responsible for early hippocampal hyperactivity seen in APP transgenic mice.
While the technique is remarkable and the demonstration that short-term application of synthetic Aβ dimers at concentrations possibly relevant to disease triggers abnormal rises in Ca2+ signaling is convincing, I have some concerns about the overall conclusion.
First, the authors used a mouse line (here, and in their previous work) that not only expresses a mutant form of human APP (APPswe) driven by the Thy1 promoter, but it also expresses a mutant form of PS1 (i.e., PS1G384A). Since the mid-1990s (Ito et al., 1994), numerous lines of evidence suggest that PS1 is involved in regulating Ca2+ homeostasis (see, for review, Green and LaFerla, 2008; Ho and Shen, 2011), and that FAD PS mutations cause abnormal Ca2+ signaling. More recently, the De Strooper and Bezprozvanny groups showed that several FAD PS1 mutations are loss-of-function mutations affecting ER Ca2+ leak activity (Nelson et al., 2007). The G384A PS1 mutant that Busche and coworkers used leads to greater than a twofold increase in ER Ca2+ concentration. This complicates interpretation. PS45 (Thy1-PS1G384A) and APP23 mice would be appropriate controls for PS-specific effects and for the γ-secretase inhibitor experiments that suggested calcium increases were due to APP/Aβ overexpression.
Second, the use of topically applied synthetic Aβ dimers in mice is somewhat counterintuitive. While Ca2+ signaling is altered in 1.5-month-old, plaque-free APP23xPS45 animals, no data are provided to determine what forms of soluble Aβ species are present in these mice. Considering that levels of soluble Aβ dimers parallel the formation of plaques (Larson and Lesne, 2012), and taking into account that young (1.5- to 1.8-month-old) bigenic mice do not display amyloid plaques (Supplementary Fig. S1), it is difficult to justify the use of synthetic dimers in this paradigm. In my opinion, it would have been more interesting to know what is present in the young bigenic mice and to try to purify and apply it to their open cranium preparation to demonstrate what is causing this putative increase in Ca2+ signaling.
Overall, I am puzzled as to how the authors conclude that the elevation of Ca2+ signaling (and by proxy, neuronal activity) observed in APP23xPS45 neurons is due to soluble Aβ.
The second study, by Chakroborty and colleagues, also deals with abnormal calcium signaling in the 3xTg AD mouse model created by Frank LaFerla’s group. These animals express mutant forms of APP (APPswe), human tau (TauP301L), and presenilin-1 (PS1M146V-KI) (Oddo et al., 2003). In particular, the authors try to address the origin of altered Ca2+ homeostasis that may underlie synaptic depression. In presymptomatic ~1.5-month-old 3xTg-AD, the upregulation of ryanodine receptor (RyR) activity appears counterbalanced by increases in presynaptic spontaneous vesicle release, altered probability of vesicle release, and upregulated postsynaptic SK channel activity. The authors conclude that ER Ca2+ disruptions due to modulation of RyR signaling are associated with PS1 mutations, rather than tau or Aβ. While this work reveals new details in dysregulation of Ca2+ homeostasis in 3xTg-AD mice, one would expect to compare 3xTg-AD with APPTau, PS1M164V-KI, and non-transgenic littermates, especially since this group previously showed that both 3xTg-AD and PS1M164V-KI mice display a similar “calciumopathy” (Stutzmann et al., 2006).
Altogether, my personal interpretation is that the new results presented here suggest that responses to RyR modulation could be solely due to mutant PS1 expression, or to some effect of human APP/Aβ/Tau expression on PS1M146V-induced calcium alterations.
In summary, I think it is extremely important to distinguish the effects of mutant PS1 on Ca2+ signaling from changes triggered by human Aβ or tau species in APPxPS1 mice in order to attribute causality and consequence between the molecules involved.
References:
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012 May 29;109(22):8740-5. Epub 2012 May 16 PubMed.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
Green KN, Laferla FM. Linking calcium to Abeta and Alzheimer's disease. Neuron. 2008 Jul 31;59(2):190-4. PubMed.
Ho A, Shen J. Presenilins in synaptic function and disease. Trends Mol Med. 2011 Nov;17(11):617-24. PubMed.
Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994 Jan 18;91(2):534-8. PubMed.
Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007 May;117(5):1230-9. Epub 2007 Apr 12 PubMed.
Larson ME, Lesné SE. Soluble Aβ oligomer production and toxicity. J Neurochem. 2012 Jan;120 Suppl 1:125-39. PubMed.
Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer's disease mice. J Neurosci. 2012 Jun 13;32(24):8341-53. PubMed.
Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006 May 10;26(19):5180-9. PubMed.
View all comments by Sylvain LesneMake a Comment
To make a comment you must login or register.