Somatic Mutations Accrue in Alzheimer's Neurons
Quick Links
As we age, our neurons steadily accrue mutations. Is this any worse in Alzheimer’s disease? Yes, according to researchers led by Christopher Walsh, Eunjung Alice Lee, and Michael Lodato at Boston Children’s Hospital. In the April 20 Nature, they reported that, compared to control cells, excitatory neurons from AD postmortem tissue carried more single-nucleotide variations. These fell into three main types: bases that had been oxidized, mutations that clustered within genes, and coding variants predicted to cripple proteins. The scientists believe that amyloid triggers a crush of oxidative stress that creates so many changes to DNA that repair enzymes become overwhelmed, allowing somatic mutations to accrue.
- In AD, somatic mutations included oxidized nucleotides and coding changes.
- The mutation load may stress DNA repair enzymes, allowing errors to pile up.
- No somatic mutations found in APP, PSEN1, PSEN2, or APOE.
“Intriguingly, the study revealed that the pattern of sSNV accumulation in AD neurons is different from normal genome aging,” wrote Ekaterina Rogaeva, University of Toronto, to Alzforum. “Notably, [the authors] did not observe somatic pathogenic mutations in AD-related genes, and suggested that the broad genomic distribution of sSNVs indicates secondary events in AD pathogenesis.”
Karl Herrup, University of Pittsburgh Medical Center, Pennsylvania, called the study a “monumental tour de force.” “The authors present an unparalleled, ultra-high-resolution look at genomic integrity across the human lifespan and its accelerated loss in Alzheimer's disease,” he wrote. Mark Mattson, Johns Hopkins University, Baltimore, wrote, “This study shows that mutations, likely caused by free radical damage, accumulate in an age-dependent manner and could lead to gene dysfunction that contributes to the disease process.”
Previously, Walsh and other researchers had seen somatic mutations accumulate in neurons during both normal aging and neurodegeneration (Lodato et al., 2018; Oct 2018 news; Jun 2004 news).
In sporadic AD, scientists had also reported somatic changes in familial AD and tauopathy genes, such as PSEN1 and 2, and MAPT (Jul 2015 news). Jerold Chun and colleagues at Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, reported genetic rearrangements in somatic cells that led to extra copies of APP harboring AD mutations, though that work was later questioned (Nov 2018 news; Jul 2020 news). Could somatic mutations throughout the genome, beyond known AD genes, contribute to the disease?
To find out, co-first authors Michael Miller and August Yue Huang sequenced the entire genomes of neurons from nine adults ages 57 to 91 who’d had AD and 20 controls aged five months to 104 years. Samples were from the Massachusetts Alzheimer’s Disease Research Center and the NIH Neurobiobank. About half of the participants were female; most were Caucasian. All AD cases were Braak stages V or VI.
The scientists isolated NeuN-positive neurons from prefrontal cortex and hippocampal tissue, then used fluorescence-activated nuclear sorting to select the largest nuclei for genome sequencing. These belonged to pyramidal, excitatory neurons, which are vulnerable to tau tangles and death in AD (Jan 2021 news; Dec 2018 conference news). The researchers measured the extent and types of somatic, single-nucleotide variants (sSNVs) in 120 neurons from AD cases and in 199 from controls.
AD neurons accumulated more damage throughout their genome than neurons from age-matched controls. Normal aging added about 20 sSNVs per neuron each year, while AD had spurred hundreds more mutations than expected, amounting to more than a decade of excess DNA damage.
Mutation load varied widely among neurons and between brain regions in AD, highlighting the heterogeneity of the disease (see image below). “The damage done in one cell is unlikely to be precisely mirrored in any other cell. Like snowflakes, no two degraded genomes are exactly alike,” noted Herrup.
Hippocampal neurons carried more variants than cortical ones, on average, but cortical neurons were more likely to be highly mutated. Why would that be? Because amyloid plaques and tau tangles in the hippocampus appear to precede prefrontal cortex damage, the authors note, they think heavily mutated hippocampal neurons had already perished, making that area seem less burdened by DNA damage at autopsy.
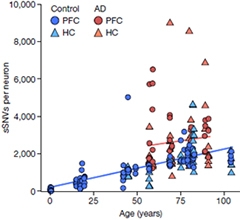
Aging Bad, AD Worse. Somatic single-nucleotide variations (sSNVs) accrue in neurons from the prefrontal cortex (circles) and hippocampus (triangles) during normal aging (blue) and even more so in AD (red). A few diseased hippocampal neurons had the highest mutation burden. [Courtesy of Miller et al., Nature, 2022.]
The types of sSNVs the scientists saw were not the same between healthy controls and AD cases. Healthy neurons tended to swap cytosine for thymine or vice versa, while AD neurons had more cytosine to adenosine changes and more 8-oxoguanine. The latter is generated by oxidative damage, which can run rampant in AD (Pao et al., 2020; Gabbita et al., 1998; Apr 2012 news). An unrepaired 8-oxoG may become a C>A mutation because DNA polymerase might misread the damaged G as a T and then incorporate a matching A on the complementary strand. The researchers found more C>A mutations in transcribed than in untranscribed DNA strands, suggesting that transcription-coupled nucleotide excision repair may falter. In keeping with this, the AD neurons also had more T>A variants, which are also a product of faulty excision repair. All told, the signature of mutations in AD differed from those in age-matched controls.
Could these mutated transcripts create faulty proteins? Compared to controls, AD neurons had more amino-acid-altering mutations. The authors thought these could make dysfunctional proteins or neoantigens that may activate T-cells to patrol the brain, as noted in AD previously (Jan 2020 news). The scientists also predicted that the higher the mutation load rose in AD, the likelier a harmful mutation would befall both alleles of the same gene, effectively knocking out the corresponding protein.
Were known AD risk genes among the ones bearing mutations? No pathogenic mutations in APP, PSEN1, PSEN2, or APOE emerged. This contradicts the previous finding that PSEN1, PS2, and MAPT mutations occur in somatic cells in sporadic AD. In a separate study, researchers led by Chunyu Liu of SUNY Upstate Medical University, Syracuse, New York, also found no sSNVs within coding regions of APP, PSEN1, PSEN2, or APOE when they analyzed whole-genome, whole-exome, and RNA-Seq data (Min et al., 2021). However, Liu, and first author Shishi Min cautioned against overinterpreting the data. “Given that all the studies have only sequenced very small numbers of AD patients, the generalizability of the findings must be read with caution. [The mutation signatures] might be just incidental in a few individuals,” they wrote (comment below).
Are somatic mutations a cause or an effect of AD pathology? Walsh and colleagues believe it’s the latter. They propose that reactive oxygen species generated in the course of the disease process may speed oxidative nucleotide damage and overwhelm repair enzymes, allowing mutations to accelerate (see image below). This may happen in other neurodegenerative diseases, as well (Apr 2022 news; Apr 2007 news).

The Oxidative Cascade? Aβ oligomers outside neurons spur tau tangles and reactive oxygen species within. The continuous assault damages DNA and overwhelms repair mechanisms, such as nucleotide excision repair. Single-nucleotide variations slip through and persist as somatic mutations, leading to neuron death. [Courtesy of Miller et al., Nature, 2022.]
Mattson thinks the authors’ take made sense. “Amyloid aggregating on the membrane of neurons causes lipid peroxidation and oxidative stress, so the sSNVs could be an intermediate between free radicals and neurodegeneration,” he said (reviewed by Butterfield, 2020). For his part, Herrup thought the relationship could go either way, noting that the higher mutation rate may begin decades before symptoms. “It is just as likely, given the timing, that the sSNVs and their downstream consequences initiate the events leading to tau and amyloid deposits,” he wrote.
Do these somatic mutations play a critical part in the demise of neurons in AD? Huntington Potter, University of Colorado Alzheimer’s and Cognition Center, Denver, doesn’t think so. “The very low numbers of sSNVs per neuron, and of potential neurons with a critical gene knocked out, makes it unlikely that these defects alone could lead to the extensive neuron loss observed in AD,” he wrote (full comment below). Mattson suggested comparing the sSNV rate among neurons that are vulnerable to AD and those that are not. “If there’s a role for these somatic mutations in neuronal dysfunction and degeneration, then they shouldn’t be as prevalent in neurons from resilient brain regions,” he said. Ekaterina Rogaeva, University of Toronto, wondered if a high somatic mutation load is a general phenomenon among neurons in different brain regions affected by other neurodegenerative diseases (full comment below).
More broadly, somatic mutations do extend beyond excitatory neurons. Scientists led by Manolis Kellis at MIT found that glia from AD brain tissue were also chock-full of damaged DNA. In a new study, co-first authors Maria Kousi and Carles Boix compared whole-genome and RNA sequencing data from 4,014 cells taken from the prefrontal cortices of 19 people who had had AD and from 17 controls. AD cells harbored more somatic mutations than controls, with excitatory neurons, astrocytes, oligodendrocytes, and disease-associated senescent cells having the most. In this sample, damaged genes fell into disease-relevant pathways, including neuronal energy regulation, endocytic trafficking, lipid metabolism, and proteostasis. Their manuscript was recently uploaded to bioRxiv (Kousi et al., 2022).—Chelsea Weidman Burke
References
News Citations
- Islands of Mutated Neurons Dot the Brain. Are They Bad for Us?
- After 40, DNA Damage Accrues in Genes, Hampering Expression
- Could Genetic Mosaicism in Adult Neurons Precipitate Disease?
- Could Rogue APP Variants Invade Genome of Individual Neurons?
- Rogue APP Claim Embroiled in Contamination Concerns
- Selective Vulnerability News: RORB Neurons Are First Victims of Tangles
- Tau Silences, Aβ Inflames; Hitting Excitatory Synapses Hardest
- Evidence Links Aging, Oxidative Stress, and AD Pathology
- Attack of the Clones? Memory CD8+ T Cells Stalk the AD, PD Brain
- DNA Nuclease Fans the Flames of Huntington’s Disease
- A Growing Problem: Oxidative Damage Drives Trinucleotide Expansions in Aging Brain
Paper Citations
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, Yang P, Chittenden TW, Hatem NE, Ryu SC, Woodworth MB, Park PJ, Walsh CA. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018 Feb 2;359(6375):555-559. Epub 2017 Dec 7 PubMed.
- Pao PC, Patnaik D, Watson LA, Gao F, Pan L, Wang J, Adaikkan C, Penney J, Cam HP, Huang WC, Pantano L, Lee A, Nott A, Phan TX, Gjoneska E, Elmsaouri S, Haggarty SJ, Tsai LH. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer's disease. Nat Commun. 2020 May 18;11(1):2484. PubMed.
- Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998 Nov;71(5):2034-40. PubMed.
- Min S, Li Z, Shieh A, Giase G, Bao R, Zhang C, Kuney L, Kopp R, Asif H, Alliey-Rodriguez N, Qin L, Craig DW, Faulkner GJ, Gershon ES, Tang B, Chen C, Liu C. Absence of coding somatic single nucleotide variants within well-known candidate genes in late-onset sporadic Alzheimer's Disease based on the analysis of multi-omics data. Neurobiol Aging. 2021 Dec;108:207-209. Epub 2021 Jul 21 PubMed.
- Butterfield DA. Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories. Ageing Res Rev. 2020 Mar 20;:101049. PubMed.
- Kousi M, Boix C, Park YP, Mathys H, Sledzieski S, Peng Z, Bennett DA, Tsai LH, Kellis M. Single-cell mosaicism analysis reveals cell-type-specific somatic mutational burden in Alzheimer’s Dementia. bioRxiv. April 22, 2022 bioRxiv
Further Reading
Primary Papers
- Miller MB, Huang AY, Kim J, Zhou Z, Kirkham SL, Maury EA, Ziegenfuss JS, Reed HC, Neil JE, Rento L, Ryu SC, Ma CC, Luquette LJ, Ames HM, Oakley DH, Frosch MP, Hyman BT, Lodato MA, Lee EA, Walsh CA. Somatic genomic changes in single Alzheimer's disease neurons. Nature. 2022 Apr;604(7907):714-722. Epub 2022 Apr 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Toronto
The study by Miller et al. is very interesting. It investigated single-cell, whole-genome sequencing data from 319 excitatory neurons of AD cases and controls. The results confirmed that somatic mutations accumulate during aging; and demonstrated that neurons from AD-affected tissues (prefrontal cortex and hippocampus) showed a significant increase in single-nucleotide variants vs. age-matched controls. Notably, Miller, et al. did not observe somatic pathogenic mutations in AD-related genes, and suggested that the broad genomic distribution of sSNVs indicates secondary events in AD pathogenesis (e.g., DNA damage by reactive oxygen species).
Intriguingly, the study revealed that the pattern of sSNV accumulation in AD neurons is different from normal genome aging (mutations classified as signature A). AD neurons show an increase in signature C, which includes C>A substitutions known to be associated with oxidative damage. Notably, the burden of AD-linked signature C shows more variation between neurons than that for aging-linked signature A. Likely, signature A and signature C arise from different mechanisms. It remains to be clarified if genomic damage linked to signature C results from AD-related defects in DNA repair. Also, it is unknown if the observed association is a cause or a consequence of neurodegeneration.
As for any genetic study, validation in an independent dataset is needed. The result is highly variable, even between neurons from the same individual, and the investigated cohort is modest in size. The study included 11 aged controls and eight AD cases at a late disease stage (Braak V-VI) with the number of investigated neurons ranging between three to nine per case. Intriguingly, for some cases the excess of sSNVs per neuron was as expected for the age, e.g., in case #4522.
A major question that remains is if the observed association is specific to AD or a general phenomenon for other age-related neurodegenerative disorders. This is of note, because neurodegenerative diseases share overlapping clinical and pathological features and are caused by the deterioration and loss of neurons, with different brain regions affected in specific diseases.
University of Pittsburgh School of Medicine
The monumental study of Miller et al. is a tour de force. Using whole-genome sequencing of the genomes of single excitatory neurons, the authors present an unparalleled, ultra-high-resolution look at genomic integrity across the human lifespan and its accelerated loss in Alzheimer's disease. They show a clear, linear increase in Single Nucleotide Variants with age. They also show that excitatory neurons in persons with Alzheimer's display more evidence of DNA damage than would be expected based on age alone.
Probing more deeply, they find certain types of somatic variants predominate over others in ways that are highly suggestive of oxidative DNA damage as a major contributor to the increase. None of these relationships is unexpected based on prior work, but the level of detail provided in this study is remarkable and the clarity of the findings dramatically increases confidence in our models of aging and its link to Alzheimer's disease. Without a doubt the dataset will be a valuable resource to bioinformaticians for years to come.
While many questions are answered by the study, others are raised. Many of these boil down to the all-too-familiar question of which way do the lines of causality run. Stated simply: Does the disease cause the DNA damage or does the DNA damage cause the disease? The authors themselves ask about the linkage between the proteinopathies involving tau and Aβ and the increase in SNVs. In truth, this question could be asked in either direction as it is just as likely, given the timing, that it is the SNVs and their downstream consequences that initiate the events leading to tau and amyloid deposits. In any of the figures that plot damage as a function of age, extending the AD line to the left to find its intersection with the line formed by the neurotypical cells suggests that the difference in SNVs has its onset well back into a person's youth. The Scottish IQ (Whalley et al., 2000) and Nun Studies (Snowdon et al., 1996) spring to mind.
There are a few technical concerns that are worth keeping in mind as we celebrate the triumph of this work. Despite the volume of data generated, we are only looking at neurons, and only at excitatory neurons. Do the rates of aging in different cell types parallel each other? Would "Signature C," defined as more disease-specific, have its homologs in other brain cell types? The sequencing protocols are designed to detect SNVs. Small deletions or amplifications, however, might well have been missed, especially since an effort was made during the amplification step to use only shorter genomic fragments to build the libraries.
My own lab has shown that inhibiting DNA repair can lead to small double-stranded genomic fragments being exported to the cytoplasm (Song et al., 2019; Song et al., 2021). In microglia, where the cGAS/STING signaling system is present, these fragments trigger a sterile inflammatory response. Yet similar cytoplasmic DNA fragments are found in neurons (Song et al., 2019), suggesting the loss of nuclear DNA occurs in them, as well.
A final feature of the data that stands out from figure to figure is how much the variance increases with Alzheimer's disease. The authors comment on this but do no in-depth analysis. It reminds us that each cell's genome is unique, and the damage done in one cell is unlikely to be precisely mirrored in any other cell. Like snowflakes, no two degraded genomes are exactly alike.
These minor concerns aside, the authors deserve a huge shout-out for the scale of the work they undertook and the intelligent and comprehensive way in which they have approached and analyzed their data.
References:
Snowdon DA, Kemper SJ, Mortimer JA, Greiner LH, Wekstein DR, Markesbery WR. Linguistic ability in early life and cognitive function and Alzheimer's disease in late life. Findings from the Nun Study. JAMA. 1996 Feb 21;275(7):528-32. PubMed.
Song X, Ma F, Herrup K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J Neurosci. 2019 Aug 7;39(32):6378-6394. Epub 2019 Jun 12 PubMed.
Song X, Aw JT, Ma F, Cheung MF, Leung D, Herrup K. DNA Repair Inhibition Leads to Active Export of Repetitive Sequences to the Cytoplasm Triggering an Inflammatory Response. J Neurosci. 2021 Nov 10;41(45):9286-9307. Epub 2021 Sep 30 PubMed.
Whalley LJ, Starr JM, Athawes R, Hunter D, Pattie A, Deary IJ. Childhood mental ability and dementia. Neurology. 2000 Nov 28;55(10):1455-9. PubMed.
SUNY Upstate Medical University
Central South University
This new paper by Miller et al. on somatic mutations in AD neurons is interesting. We are very glad to see that our findings of absence of coding somatic mutations in known AD risk genes APP, PSEN1, PSEN2 or APOE (Min et al., 2021) is replicated by this new study with additional samples and about three times as many neurons sequenced.
Another major highlight of this new paper is the signature C enriched in the AD brains. However, we are a little bit skeptical about this exact finding. We analyzed mutational signatures in our bulk tissue DNA data as reported in our paper above. We did not detect such a signature. We also performed a similar analysis on our single-cell WGS data and failed to detect case-control differences for both an age-related signature and another signature related to oxidative DNA damage. We had a hard time getting this negative result published.
We note that another paper (Park et al., 2019) reported a mutational signature (SBS18) in their bulk DNA sequencing results that was different from this new paper by Miller et al. (SBS8), and was also different from our own results. This leads us to think that the reported mutational signatures might not be robust findings for AD neurons. They might be incidental changes in a few individuals.
We also noticed that the authors' primary data supporting the case-control difference in signature C contribution pooled all neurons of all individuals together (Fig. 2e). It could be checked whether those signature C-loaded neurons were from a small number of individuals or from most of the samples. Given that all the studies have only sequenced a very small numbers of AD patients, the generalizability of the findings needs to be read with caution.
We agree with Miller et al. that there might be some oxidative DNA damage in some of the AD brains. But this could be associated with any of many forms of mutational signatures, not necessarily signature C. Moreover, how common such damage is in AD patient brains remains to be tested in large samples.
References:
Min S, Li Z, Shieh A, Giase G, Bao R, Zhang C, Kuney L, Kopp R, Asif H, Alliey-Rodriguez N, Qin L, Craig DW, Faulkner GJ, Gershon ES, Tang B, Chen C, Liu C. Absence of coding somatic single nucleotide variants within well-known candidate genes in late-onset sporadic Alzheimer's Disease based on the analysis of multi-omics data. Neurobiol Aging. 2021 Dec;108:207-209. Epub 2021 Jul 21 PubMed.
Park JS, Lee J, Jung ES, Kim MH, Kim IB, Son H, Kim S, Kim S, Park YM, Mook-Jung I, Yu SJ, Lee JH. Brain somatic mutations observed in Alzheimer's disease associated with aging and dysregulation of tau phosphorylation. Nat Commun. 2019 Jul 12;10(1):3090. PubMed.
University of Colorado Alzheimer’s and Cognition Center
The data presented here are the result of carefully designed experiments using two different single-cell whole-genome sequencing methods. The important conclusion is that brain neurons in people with Alzheimer’s disease accumulate numerous somatic single-nucleotide variations, which are different from another set of sSNVs that were shown previously to accumulate in brain neurons during normal aging.
The implication is that AD neurons accumulate a specific somatic mutational signature that could negatively impact their function and survival. The potential significance is increased because the AD-specific sSNVs are the same types that can be attributed to oxidative stress. Because oxidative stress has been shown to occur in the brain in AD and other neurodegenerative diseases (Gabbita et al., 1998), this connection between a physiological pathology and a disease-specific mutational signature of genomic damage suggests an approach to developing novel disease interventions based on preventing oxidative stress and/or its consequent DNA damage.
The research team also found that the AD-specific sSNVs were associated with sites of transcription, suggesting that they may preferentially affect the function of important neuronal genes. The authors modeled the potential effects of sSNVs that are in close proximity to each other and are sufficiently numerous to lead to neurons that have both copies of an important gene knocked out (~0.4 percent), which is much higher than might be expected from the approximately 1,000 total sSNVs that are estimated to occur per neuron.
Although the rigorous approach and data presented in the paper are convincing, the very low numbers of sSNVs per neuron and of potential neurons with a critical gene knocked out, which might not all survive for the analyses, makes it appear unlikely that these defects alone could lead to the extensive brain neuron loss observed in AD compared to normal aging. If AD-specific sSNVs are caused by oxidative stress, then the sSNVs would be a downstream step in the AD pathogenic pathway that would need to be further amplified to result in massive neuronal cell loss.
Another important impact of the paper is its reinforcement of a concept that developed from previous studies—that many neurons in the brains of people with AD accumulate genomic defects. The most striking previous finding is that brain neurons, and also non-neuronal cells, in AD and frontotemporal dementia, exhibit mosaic aneuploidy that encompasses 10–30 percent of neurons at autopsy, as measured by various single-cell assessments, including DNA in situ hybridization (Granic et al., 2010; Caneus et al., 2018; Arendt et al., 2010; for review see Potter et al., 2019). Such chromosome aneuploidy would lead to massive imbalances in gene expression and has been shown to induce preferential apoptosis, potentially accounting for 90 percent of the neuronal loss found in end-stage AD (Arendt et al., 2010). Although single-cell DNA sequencing has often not detected such aneuploidy (Knouse et al., 2014; Lee et al., 2018), the most likely reason is that the quality control algorithms commonly used reject the data from more than half the cells, including all apoptotic cells, thus undercounting aneuploid neurons. Although controversial, genomic mosaicism in the AD brain due to the accumulation of cells that have gained an extra copy of the APP gene has also been reported (Lee et al., 2018; Kim et al., 2020; Lee et al., 2020).
The take-home message of all of these studies is that the genomic integrity of the brain we are born with is not permanent, and protecting it from genomic damage is a promising approach to preventing neurodegenerative diseases and even cognitive decline during "normal" aging.
References:
Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998 Nov;71(5):2034-40. PubMed.
Granic A, Padmanabhan J, Norden M, Potter H. Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol Biol Cell. 2010 Feb 15;21(4):511-20. PubMed.
Caneus J, Granic A, Rademakers R, Dickson DW, Coughlan CM, Chial HJ, Potter H. Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol Biol Cell. 2018 Mar 1;29(5):575-586. Epub 2017 Dec 27 PubMed.
Arendt T, Brückner MK, Mosch B, Lösche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010 Jul;177(1):15-20. PubMed.
Potter H, Chial HJ, Caneus J, Elos M, Elder N, Borysov S, Granic A. Chromosome Instability and Mosaic Aneuploidy in Neurodegenerative and Neurodevelopmental Disorders. Front Genet. 2019;10:1092. Epub 2019 Nov 7 PubMed.
Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A. 2014 Sep 16;111(37):13409-14. Epub 2014 Sep 2 PubMed.
Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, Chun J. Somatic APP gene recombination in Alzheimer's disease and normal neurons. Nature. 2018 Nov;563(7733):639-645. Epub 2018 Nov 21 PubMed.
Kim J, Zhao B, Huang AY, Miller MB, Lodato MA, Walsh CA, Lee EA. APP gene copy number changes reflect exogenous contamination. Nature. 2020 Aug;584(7821):E20-E28. Epub 2020 Aug 19 PubMed.
Lee MH, Liu CS, Zhu Y, Kaeser GE, Rivera R, Romanow WJ, Kihara Y, Chun J. Reply to: APP gene copy number changes reflect exogenous contamination. Nature. 2020 Aug;584(7821):E29-E33. Epub 2020 Aug 19 PubMed.
Tel Aviv University
Our study, cited below (Ivashko-Pachima et al., 2019), completely agrees with the new exciting study on somatic mutations in Alzheimer's neurons. For further corroboration of somatic mutations in Alzheimer’s brains, see this recent study cited below (Soheili-Nezhad et al., 2020).
Our studies also discovered mutations in glial cells, which will impact neuro-glial interactions. Specifically, we found an enrichment of high-impact somatic mutations in cytoskeletal-associated proteins, directly driving tauopathy, and advocating for Tau-targeted Alzheimer’s treatment.
Furthermore, key genes that undergo mutations in Alzheimer’s disease brains are long genes, also prone to de novo mutations driving autism and intellectual disabilities. For example, activity-dependent neuroprotective protein (ADNP) presents de novo mutations leading to the autistic ADNP syndrome as well as somatic mutations driving tauopathy in the Alzheimer’s brain. Importantly, the ADNP snippet and drug candidate NAP (Davunetide) directly protects against tauopathy as well as oxidative stress (Jul 2002 news).
Regarding competing financial interests, I am chief scientific officer of ATED Therapeutics Ltd.
References:
Ivashko-Pachima Y, Hadar A, Grigg I, Korenková V, Kapitansky O, Karmon G, Gershovits M, Sayas CL, Kooy RF, Attems J, Gurwitz D, Gozes I. Discovery of autism/intellectual disability somatic mutations in Alzheimer's brains: mutated ADNP cytoskeletal impairments and repair as a case study. Mol Psychiatry. 2019 Oct 30; PubMed.
Soheili-Nezhad S, van der Linden RJ, Olde Rikkert M, Sprooten E, Poelmans G. Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer's disease: Implications for disease etiology. Alzheimers Dement. 2021 Mar;17(3):489-499. Epub 2020 Oct 19 PubMed.
Make a Comment
To make a comment you must login or register.