Sought FUS, Found TAF—A New Fibril in Frontotemporal Dementia
Quick Links
While filaments of either tau or TDP-43 underlie 90 percent of frontotemporal dementia cases, the proteopathic culprits behind the remaining 10 percent have eluded researchers. Fused in sarcoma (FUS) often forms inclusions in such cases, hence Benjamin Ryskeldi-Falcon and colleagues at the MRC Laboratory of Molecular Biology, Cambridge, U.K., expected to find FUS fibrils when they put samples from these patients' brains through the cryoEM ringer. As reported December 6 in Nature, they happened upon filaments of another RNA-binding protein, namely TAF15, the TATA-box binding protein-associated factor 2N. Nary a fibril of FUS was to be found, although FUS comingled with TAF15 in inclusions. The core of the TAF15 filaments comprised a 13-β-strand fold from the protein’s low-complexity domain.
- Cryo-EM reveals TAF15 filaments in frontal and motor cortices of four people with FTLD.
- The fibril core comprises 13 β-sheet strands.
- Scientists spotted FUS in protein inclusions, but no FUS fibrils.
The study presents TAF15 as a distinct proteinopathy underlying some cases of FTLD. TAF15 joins, in infamy, Aβ, tau, α-synuclein, and TDP-43, the proteins known thus far to form amyloid filaments in neurodegenerative diseases.
Rare mutations in FUS cause familial amyotrophic lateral sclerosis, a disease that sometimes co-occurs with FTLD and lies on the same neuropathological spectrum (Feb 2009 news). Inclusions chock-full of the protein form in people who have FTLD without TDP-43 or tau aggregates, the other major proteinopathies in ALS/FTD (Neumann et al., 2009). However, FUS is not by itself in these inclusions. It keeps company with the related RNA binders Ewing sarcoma (EWS) and TAF15, leading some researchers to designate the disease “FTLD-FET” (Neumann et al., 2011). FUS, EWS, and TAF15—FET, for short—all can form fibrils in vitro, but until now, no filaments had been resolved from the human brain (Sep 2017 news; Sun et al., 2021; Chen et al., 2022).
First author Stephan Tetter and colleagues searched for filaments in the prefrontal and temporal cortices of four people with FTLD-FET. All three FET proteins, along with the nuclear import receptor transportin-1, appeared in insoluble brain extracts, suggesting they aggregated together in some way. Alas, cryo-EM images of more than 100,000 fibril segments from each of the four samples were compiled to reveal that TAF15 alone twisted into filaments.

TAF15 Fold. TAF15 filaments (left, yellow arrows) featured a single protofilament. The same core protofilament structure emerged in brain samples from four different people with FTLD-FET (right). [Courtesy of Tetter et al., Nature, 2023.]
The protofilament molecules of TAF15 twist to the left as they stack together to form a helix. The ordered core comprises residues 7-99 of the protein’s low-complexity domain (LCD). Perpendicular to the helical axis, each protofilament forms 13 β-strands that contort into a shape that vaguely resembles a Vespa scooter, with its handlebars formed by the N- and C-termini.
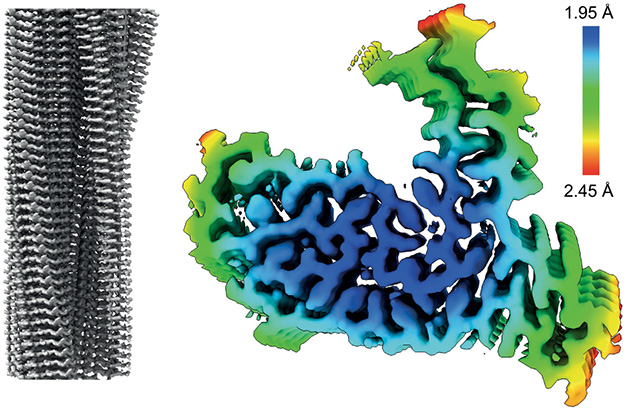
Stack of Scooters. A TAF15 filament comprises a single protofilament stack of molecules twisting left around the helical axis (left). Perpendicular to the filament axis, the protofilament core comprises 13 β-strands folded into the shape of a scooter (right).
In a configuration typical of amyloids, the β-strands form parallel β-sheets that interact with those of neighboring TAF15 molecules. Eight of the β-strands zip together with interdigitating side chains. The protofilament cores were loaded with glycine, tyrosine, and glutamine residues, which facilitated tight turns between β-sheets, stacking interactions, and networks of hydrogen bonds, respectively.
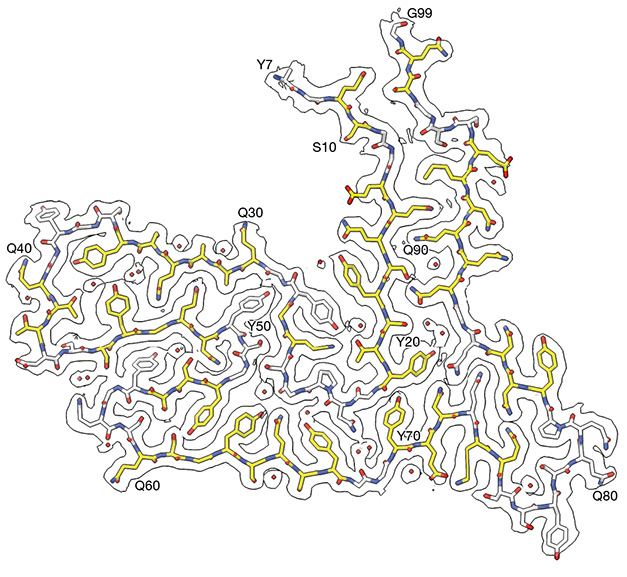
The Vespa Fold? The core protofilament structure of TAF15 fibrils includes 13 β-strands from residues 7-99 of TAF15’s LCD. The structure resembles a Vespa, with tyrosine 7 at the N-terminus and glycine 99 at the C-terminus forming “handlebars.” [Courtesy of Tetter et al., Nature, 2023.]
FET-filled inclusions have been spotted in ALS, as well. Might TAF15 form the same filaments in this disease? Two of the four donors in this study had evidence of lost myelin and neurons in their upper and lower motor neurons, as well as inclusions filled with FUS and TAF15 in their motor cortices, brainstems, and spinal cords. In fact, one had been initially diagnosed with probable ALS, then with FTLD. When the researchers teased apart motor cortex and medulla samples from these two patients, they again identified filaments of TAF15, but not FUS. The protofilament cores folded into the same configuration as those found in the prefrontal and temporal cortices.
Healthy control tissue had no TAF15 filaments, suggesting these amyloids were related to disease. This stands in contrast to another surprise filament in FTLD. Last year, Ryskeldi-Falcon’s and two other groups identified TMEM106b filaments in FTLD and in other neurodegenerative diseases (Apr 2022 news). These filaments proved to be related to aging rather than neurodegenerative disease, cropping up in brain samples from neurologically normal controls over the age of 50. In keeping with this, Tetter identified TMEM106b filaments in three of the FTLD-FET samples used in the current study. The one sample without TMEM106b fibrils came from a person who had died at the age of 30.
The group of Ian Mackenzie of the University of British Columbia in Vancouver first spotted and characterized inclusions of FET proteins in FTLD. To Mackenzie's mind, the new findings are interesting but not entirely unexpected. Even back in 2011, when Mackenzie and colleagues coined the term FTLD-FET, there were hints that TAF15 was more than a mere bystander, he wrote to Alzforum. Of the three FET proteins, TAF15 was the most insoluble, the most abundant in the inclusions, and more of it shifted from the nucleus to the cytoplasm in inclusion-bearing cells.
Ryskeldi-Falcon told Alzforum that his lab is studying why TAF15 forms fibrils, and how the fibrils influence the disease process. He believes that because TAF15 binds FUS and some of its RNA partners, it pulls FUS and other proteins, such as transportin-1, into inclusions.
Ryskeldi-Falcon said it’s possible that FUS forms fibrils, for example in rare cases of ALS caused by FUS mutations. In-depth analyses could tease out whether mutations in TAF15 might promote FTLD, and see if TAF15 filaments occur more broadly, for example in sporadic cases of ALS.—Jessica Shugart
References
News Citations
- New Gene for ALS: RNA Regulation May Be Common Culprit
- Out of Chaos, Order: Reversible Amyloid Structure Seen in Phase Separation
- Surprise! TMEM106b Fibrils Found in Neurodegenerative Diseases
Paper Citations
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009 Nov;132(Pt 11):2922-31. PubMed.
- Neumann M, Bentmann E, Dormann D, Jawaid A, Dejesus-Hernandez M, Ansorge O, Roeber S, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ang LC, Bilbao J, Rademakers R, Haass C, Mackenzie IR. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011 Sep;134(Pt 9):2595-609. PubMed.
- Sun Y, Zhang S, Hu J, Tao Y, Xia W, Gu J, Li Y, Cao Q, Li D, Liu C. Molecular structure of an amyloid fibril formed by FUS low-complexity domain. iScience. 2022 Jan 21;25(1):103701. Epub 2021 Dec 27 PubMed.
- Chen J, Yuan X, Wei P, Wang D, Chen C, Guo Q, Luo SZ, Chen L. The SGYS motif of TAF15 prion-like domain is critical to amyloid fibril formation. Biophys J. 2022 Jul 5;121(13):2613-2623. Epub 2022 May 28 PubMed.
Further Reading
Papers
- Lee M, Ghosh U, Thurber KR, Kato M, Tycko R. Molecular structure and interactions within amyloid-like fibrils formed by a low-complexity protein sequence from FUS. Nat Commun. 2020 Nov 12;11(1):5735. PubMed.
Primary Papers
- Tetter S, Arseni D, Murzin AG, Buhidma Y, Peak-Chew SY, Garringer HJ, Newell KL, Vidal R, Apostolova LG, Lashley T, Ghetti B, Ryskeldi-Falcon B. TAF15 amyloid filaments in frontotemporal lobar degeneration. Nature. 2024 Jan;625(7994):345-351. Epub 2023 Dec 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Stanford University
A unifying theme for neurodegenerative disorders is the abnormal accumulation of protein aggregates in the central nervous systems of affected individuals. Over the last ~20 years, the isolation of the proteinaceous building blocks of these pathological deposits has provided tremendous insight into disease mechanisms. Genetics discoveries have often gone hand in glove with the biochemical discoveries and, indeed, mutations in several of the genes encoding the aggregation-prone proteins are causative for rare familial forms of the disease. For example, APP (encodes Aβ) mutations or gene triplication cause early onset Alzheimer disease, SNCA (encodes α-synuclein) mutations cause early onset Parkinson disease, MAPT (encodes tau protein) mutations cause frontotemporal dementia, and TARDBP (encodes TDP-43 protein) mutations cause ALS. Filamentous forms of a C-terminal fragment of the TMEM106B protein have recently been discovered in the brains of older individuals and those with neurodegenerative disease. TMEM106B has been linked genetically to FTLD-TDP-43 through genome-wide association studies (GWAS, Van Deerlin et al., 2010). All of this is to say that identifying a new aggregated protein in a neurodegenerative disease is of great interest and will likely be highly informative and impactful.
For frontotemporal lobar degeneration (FTLD), cases can be divided into three major types, based on pathology (Cairns et al., 2007; Mackenzie and Neumann, 2016): 1) FTLD-Tau, which is characterized by tau inclusions; 2) FTLD-TDP-43, which is characterized by TDP-43 inclusions; and a rarer subtype, FTLD-FUS, which is characterized by FUS inclusions.
This paper by Ryskeldi-Falcon and colleagues presents TAF15 as the disease protein in FTLD-FUS cases (contrary to it being FUS, as expected). The authors isolate amyloid material from the brains of 4 FTLD-FUS patients and apply cryo-EM approaches to obtain models of the filamentous material, revealing it to be TAF15.
It had already been known that TAF15 pathology is a component of FTLD-FUS cases, and this subtype is indeed now known as FTLD-FET (for FUS, EWSR1, TAF15) (Neumann et al., 2011; Neumann and Mackenzie, 2019). FUS, EWSR1, and TAF15 are three very similar RNA-binding proteins. However, the new Cryo-EM studies and other analyses now show that it is only TAF15 but not EWSR1 or FUS (or any other amyloid) in the pathological deposits. This is very exciting and novel and of great interest. It really changes how the field will think about this disease and will launch efforts to study TAF15 and perhaps target it for therapy.
In a broader sense, this finding might help explain the different observations between FTLD-FUS (not caused by FUS mutations and FUS, EWSR1, TAF15, and transportin1 are present in pathological inclusions) and ALS-FUS (caused by FUS mutations and only FUS is present in pathological inclusions).
Many questions lie ahead. The authors hypothesize that TAF15 amyloids might sequester into pathological inclusions the proteins FUS, EWSR1, and transportin, a transport protein that recognizes these three RNA-binding proteins and shuttles them into the nucleus. To test this hypothesis, experiments in cell culture and in vitro, i.e., introducing TAF15 amyloids into cells and testing if this causes mislocalization of the other endogenous proteins, will be informative. The FET proteins are highly similar at the primary sequence level, so what makes TAF15 the one that forms amyloids more readily than the others? Would lowering levels of TAF15 or otherwise preventing its aggregation afford clinical benefit? Answering these questions will provide insight and motivate launching of drug discovery programs targeting TAF15 with ASOs or small molecule degraders as a therapy for this rare form of FTLD.
References:
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL, Ferrer I, Lladó A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, Decarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tuñón MT, Martínez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010 Mar;42(3):234-9. PubMed.
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, . Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007 Jul;114(1):5-22. PubMed.
Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 2016 Aug;138 Suppl 1:54-70. Epub 2016 Jun 15 PubMed.
Neumann M, Bentmann E, Dormann D, Jawaid A, Dejesus-Hernandez M, Ansorge O, Roeber S, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ang LC, Bilbao J, Rademakers R, Haass C, Mackenzie IR. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011 Sep;134(Pt 9):2595-609. PubMed.
Neumann M, Mackenzie IR. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2019 Feb;45(1):19-40. PubMed.
University of British Columbia
The finding for filaments composed of TAF-15 is interesting but not entirely unexpected. We initially introduced the terminology “FTLD-FUS” for these subtypes of tau/TDP-negative FTLD because FUS was the first protein component of the inclusions identified (Neumann et al., 2009); a discovery prompted by the identification of FUS mutations in ALS. However, once we determined that the inclusions also label for the other FET proteins (TAF15, EWS), we suggested that FTLD-FET might be a more appropriate designation (Neumann et al., 2011).
Even at that time, there were some hints that TAF15 might play a more central role in these conditions: (i) of the three FET proteins, TAF15 showed the greatest shift to the insoluble protein fraction, (ii) inclusions are more strongly and consistently labelled with antibodies against TAF15, and (iii) only TAF15 consistently showed loss of the normal physiological nuclear immunoreactivity in inclusion bearing cells, suggesting a possible loss of function.
As the authors indicate, an important issue is whether or not there are differences among the FTLD-FET subtypes (aFTLD-U, -NIFID, -BIBD) in the pathological filaments.
With that in mind, I would point out that although the authors of this paper have designated all four cases used in their study as being examples of aFTLD-U, the morphology and anatomical distribution of the inclusions and the associated clinical phenotypes of some of the cases (e.g. ALS) would be more consistent with either NIFID or BIBD.
Additional cryo-EM analysis of additional, well-characterized cases is needed.
One minor aspect of the study that is unclear to me is why biochemical analysis of the insoluble protein fraction does not disclose any disease-specific low-molecular-weight TAF15 band corresponding to the filament core.
References:
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009 Nov;132(Pt 11):2922-31. PubMed.
Neumann M, Bentmann E, Dormann D, Jawaid A, Dejesus-Hernandez M, Ansorge O, Roeber S, Kretzschmar HA, Munoz DG, Kusaka H, Yokota O, Ang LC, Bilbao J, Rademakers R, Haass C, Mackenzie IR. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011 Sep;134(Pt 9):2595-609. PubMed.
Make a Comment
To make a comment you must login or register.