α-Synuclein from Multiple System Atrophy Acts Like Prion in Mice
Quick Links
Suggesting that particular strains of human pathological α-synuclein propagate like prions, researchers led by Stanley Prusiner and Kurt Giles at the University of California, San Francisco, have reported that extracts from the brains of people with multiple system atrophy (MSA) seeded α-synuclein aggregation and neurodegeneration in transgenic mice. In contrast, α-synuclein from Parkinson’s disease patients had no such effect. The paper appeared August 31 in Proceedings of the National Academy of Sciences.
Toxic forms of α-synuclein are central to various synucleinopathies. They constitute the primary component of neuronal Lewy bodies in PD and of glial cytoplasmic inclusions in the much rarer MSA. Researchers recently reported that different structural “strains” of α-synuclein underlie diverse pathologies (see Jun 2015 news).
Like other amyloidogenic proteins, misfolded α-synuclein corrupts the native protein. Minuscule amounts of the nefarious version can seed pathology when injected into the brains of mice that express normal or mutated forms of human α-synuclein (see Apr 2012 news; Nov 2012 news). This behavior echoes toxic prions, which coax their normal brethren to misfold, setting off a chain reaction that corrupts prion proteins across the brain.
In a 2013, Prusiner and colleagues reported that α-synuclein from two MSA patients behaved similarly. When they injected teeny amounts of patient brain extract into mouse brain, clusters of phosphorylated α-synuclein spread from the injection sites and triggered neurodegeneration (see Watts et al., 2013). Does it matter where the α-synuclein comes from? To address this question, the researchers tested brain homogenates from 14 people who had died with MSA and six with PD. The MSA patients’ brains were riddled with glial cytoplasmic inclusions, the PD patients’ with neuronal Lewy bodies. None of the patients had harbored disease-associated α-synuclein mutations or additional copies of the gene, which increase risk for synucleinopathy.
The researchers employed a cell-based assay they had developed to quantify the templating characteristics of α-synuclein from brain samples (see Woerman et al., 2015). They precipitated the extracts with phosphotungstic acid, a procedure that enriches for insoluble forms of α-synuclein. They then mixed these samples with human embryonic kidney (HEK) cells expressing the PD-causing A53T α-synuclein mutant fused to yellow fluorescent protein (YFP). Four days later, they observed fluorescent aggregates of α-synuclein in cells treated with α-synuclein from MSA brains, whereas the cells treated with extracts from PD or control brains had only background levels. This is a much faster assay than the in vivo assays typically used to assess prion propagation.
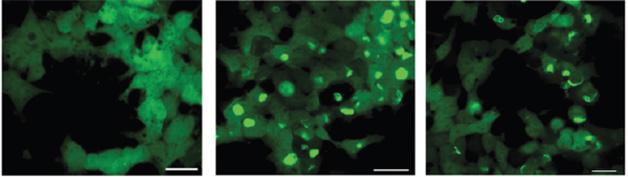
Strains Matter. When added to cells expressing fluorescentA53T α-synuclein, brain homogenates from MSA patients (middle, right), but not PD patients (left), triggered aggregation. [Courtesy of Prusiner et al., PNAS 2015.]
The researchers next tested whether the α-synuclein seeds would spread in vivo. They injected homogenates from MSA, PD, or human control brain into the thalamus of Tg83+/- mice, which express a single copy of human A53T α-synuclein. Unlike homozygous Tg83+/+ mice, Tg83+/- animals do not develop spontaneous neurodegeneration.
The results jibed with those from the HEK cells. After 100 to 150 days, mice injected with MSA-derived α-synuclein from each of the 14 MSA patients started losing coordination and turning in circles, and died two days after symptom onset. Mice injected with PD-derived α-synuclein were unaffected.
Neuropathological examination showed that neurons in mice injected with MSA extract harbored cytoplasmic and neuritic inclusions, which contained large aggregates of phosphorylated α-synuclein. These neuronal clusters were largely absent from the cortex but developed in several other non-thalamus brain regions—most prominently in the brainstem, where astrocytic gliosis was widespread. In contrast, mice injected with α-synuclein derived from PD brains harbored levels of phosphorylated α-synuclein that did not rise above those in controls. The recipient mice developed no glial inclusions, i.e., did not recapitulate the pathology distribution of the MSA patients. This was probably due to the expression pattern of the A53T transgene in the mice, according to the authors.

Prion Propagation?
Homogenates from an MSA patient triggered aggregates of phosphorylated α-synuclein (top) and activated astrocytes (bottom) in a Tg83+/- mouse. [Courtesy of Prusiner et al., PNAS 2015.]
Because serial propagation from one brain to another is a defining characteristic of prions, the researchers prepared homogenates from four of the inoculated, sick mice and transferred them into healthy Tg83+/- animals. This secondary inoculation had a similar incubation time as the first one, with mice becoming ill around 92 days later. Based on the amount of brain homogenate used for each transfer, the researchers calculated that toxic α-synuclein had amplified more than 1,000-fold with each passage.
This amplification did not occur when normal mice or transgenic mice expressing wild-type human α-synuclein served as hosts. This indicates that the A53T mutation was necessary to facilitate prion-like propagation. None of the MSA patients carried this mutation. Giles speculated that the mutation renders the protein more prone to misfolding, speeding up propagation. In an accompanying PNAS commentary, Surachai Supattapone at Dartmouth Medical School in Hanover, New Hampshire, offered an alternative explanation. He noted that in the mice, α-synuclein is expressed under control of the mouse prion promoter, which is active in neurons, while in humans with MSA the protein accumulates in glial cells. “It is possible that, to be susceptible to prion-like conformational change, WT α-synuclein must interact with one or more specific co-factors that are present in glial cells, but not neurons,” he wrote.
Propagation of human α-synuclein in mouse brains had been reported before (Recasens et al., 2014), so what makes this report more indicative of prion-like behavior? To Giles, that the strain triggered a fatal disease pins it as a prion. “When we inoculate these mice, they die,” he said.
Why didn’t α-synuclein from the brains of PD patients behave this way? Giles speculated that something about its structure must be different. That this difference is maintained when serially propagated between mice indicates that it is the α-synuclein protein itself, not something else lingering in the human brain extracts, that confers the prion-like behavior, he said.
Just because the protein spread like a prion in mice, does that mean it did so in humans? “MSA brain extracts are certainly more potent than those from PD patients,” commented Seung-Jae Lee of Seoul National University College of Medicine in South Korea. However, Lee cautioned that the data stop short of proving that these differences translate into disease mechanisms. “We don't even know whether the phenotypes we see in the inoculated mice are relevant to MSA or any other human disease,” he wrote to Alzforum.
Giles added that the consistent difference of the PD and MSA strains in both mice and cells will facilitate a deeper investigation into protein properties that confer prion activity. He plans to use the cell-based assay to scrutinize strain differences and define common mechanisms of prion activity. Supattapone added that the cell-based assay might come in handy as a diagnostic test to distinguish MSA from PD.—Jessica Shugart
References
Alzpedia Citations
News Citations
- Shape of α-Synuclein Aggregates Influences Pathology
- Toxic Synuclein Corrupts Native in Wild-Type Mice
Paper Citations
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013 Nov 26;110(48):19555-60. Epub 2013 Nov 11 PubMed.
- Woerman AL, Stöhr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, Ohyama T, Patel S, Widjaja K, Oehler A, Sanders DW, Diamond MI, Seeley WW, Middleton LT, Gentleman SM, Mordes DA, Südhof TC, Giles K, Prusiner SB. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015 Sep 1;112(35):E4949-58. Epub 2015 Aug 18 PubMed.
- Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014 Mar;75(3):351-62. Epub 2014 Feb 18 PubMed.
Further Reading
Papers
- McCann H, Cartwright H, Halliday GM. Neuropathology of α-synuclein propagation and braak hypothesis. Mov Disord. 2016 Feb;31(2):152-60. Epub 2015 Sep 4 PubMed.
Primary Papers
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015 Sep 22;112(38):E5308-17. Epub 2015 Aug 31 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Konkuk University
This paper expands on previous work in which the authors tested two MSA patient samples (Watts et a., 2013). In the current study, the authors tested samples from 12 more MSA patients and 6 PD patients and found that only the MSA brain homogenates transmit the disease to the α-synuclein transgenic mice. These are interesting data. The paper shows that the transmission of synucleinopathy is context-dependent. MSA brain extracts are certainly more potent than those from PD patients. This doesn't necessarily prove the strain-disease relationship since we don't even know whether the phenotypes we see in the inoculated mice are relevant to MSA or to any other human diseases. One caveat is that the anti-phospho-α-Syn antibody may have non-specific interactions with other proteins. Pathological examination with antibody to total alpha-synuclein might help validate the results. The million-dollar question is whether the pathology resulted from the seeded aggregation of α-synuclein.
Make a Comment
To make a comment you must login or register.