Toward an AD CRISPR Therapy: Tweaking APP Terminus Cuts Plaque
Quick Links
When in the clutches of β-secretase, the amyloid precursor protein (APP) lives up to its name, churning out the starting material for Aβ peptides and all that comes from them. What if this whole fiasco could be avoided by keeping the two apart? A manuscript posted on bioRxiv June 9 gives credence to this strategy.
- Delivered by a virus, CRISPR pulled an endosomal targeting motif off APP in mouse brain.
- This shifted APP processing away from β-secretase cleavage.
- Plaque growth, lysosomal dysfunction, gliosis, and functional decline slowed for months.
Researchers led by Subhojit Roy at the University of California, San Diego, used CRISPR gene editing to remove a six-residue motif that targets APP to the endosome, where β-secretase, aka BACE1, lies in wait. In APP knock-in mice infected with a virus carrying CRISPR machinery, APP processing shifted from this amyloidogenic path toward α-secretase cleavage instead. Predictably, this reduced the Aβ plaque burden, inflammation, synaptic deficits, and memory loss.
The authors believe this gene therapy approach holds promise for people with familial, or even sporadic, Alzheimer’s disease. Participants of the dominantly inherited Alzheimer’s Network, aka DIAN, have been clamoring for gene therapy approaches to fix their disease; this approach represents one such attempt.
“A universal one-and-done gene therapy for AD, which the authors have set as their long-term goal, is alluring, especially in a field that has struggled for decades to find a suitable therapy that will stop the disease process in its tracks,” wrote Justyna Dobrowolska Zakaria of Northwestern University in Chicago. Still, she said a lot more work needs to be done before bringing the gene-editing therapy to the clinic, starting with perfecting delivery into the human brain.
While Aβ-targeted therapies can mop up existing aggregates, other strategies have sought to curb Aβ production. One such approach, BACE1 inhibition, fell flat in trials due to cognitive side effects, though some scientists propose trying again with lower drug doses (Jul 2023 conference news). Another approach is to quash translation of APP with antisense nucleotides, though that comes at the expense of its physiological function (Nov 2023 conference news). Roy’s approach avoids these pitfalls by keeping BACE and APP apart. Previously, the scientists had reported that clipping off the final exon of APP in human neurons yielded a truncated protein that lingered on the cell surface, where it was mostly processed by α-secretase (Sun et al., 2019). The exon contains the YENPTY motif that guides the precursor to endosomes.
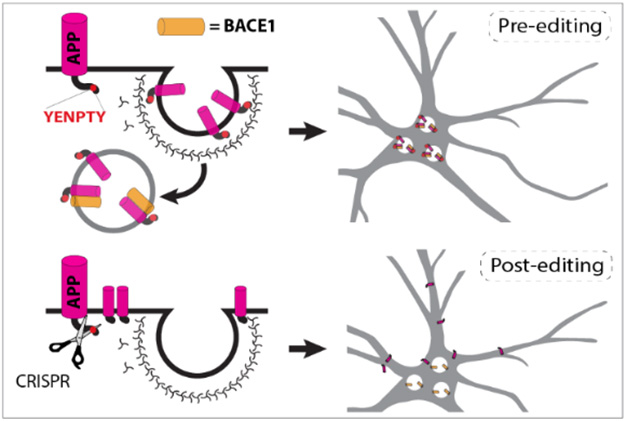
Keep Away. The YENPTY motif at APP’s C-terminus routes it to the endosome, where it is exposed to BACE1 cleavage (top). After CRISPR editing, APP stays on the cell surface, safely apart from BACE1 (bottom). [Courtesy of Aulston et al., bioRxiv, 2024.]
In their new study, first author Brent Aulston and colleagues tested this in APPNL-G-F knock-in mice, which start to develop plaques by 2 months of age. They crossed these to mice expressing Cas9 in the brain, the endonuclease that carries out CRISPR’s DNA snipping. To target APP’s final exon, the scientists packaged a guide RNA into an AAV vector optimized to cross the mouse blood-brain barrier.
After Aulston injected it into the blood, the virus broadly infected the brain. Editing worked with 90 percent efficiency in neurons, where most of the APP lacked the final 18 amino acids. Levels of sAPPα, a soluble product of α-secretase, were 25 percent higher, while sAPPβ was 25 percent lower than in mice treated with a nonspecific guide RNA. This suggested that the editing indeed sequestered APP from BACE1.
How about Aβ peptides? Ten months after infection, levels of insoluble Aβ42 and Aβ40 were half those in controls. This translated into less plaque. The difference grew over time as APPNL-G-F mice continued to lay down amyloid while APP-edited mice did so more slowly (image below).
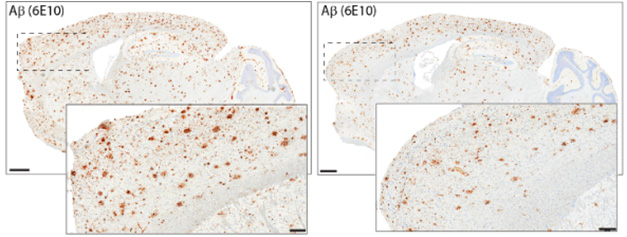
Avoid Amyloidosis. Ten months after APP-KI/Cas9 mice were infected with virus carrying an APP-editing guide RNA, they had less amyloid plaque (right) than did mice infected with a control guide RNA (left). [Courtesy of Aulston et al., 2024.]
Cutting off APP’s last exon countered disease phenotypes in the APP-KI mice. Enlarged lysosomes, microgliosis and astrocytosis, and synaptic function were less pathological, as was memory loss gauged by how long mice explored a novel object.
Roy thinks the benefits of this editing might stem not only from Aβ reduction. There’s also a drop in β-CTF, a β-secretase product reported to exacerbate lysosomal dysfunction (Aug 2019 news). A study from Ralph Nixon’s lab at New York University claims the YENPTY motif in β-CTF derails lysosomal acidification (Jul 2023 news).
In other experiments, Roy’s group edited APP in the knock-in mice from birth. This almost completely prevented formation of Aβ plaques, particularly in mice expressing only one copy of the NL-G-FAPP variant, i.e., mice that better reflect the heterozygous nature of most familial AD mutations. Single-nucleus transcriptomic analysis in these mice indicated that their microglia remained in a homeostatic state at 10 months of age, at which point these cells had transitioned into a disease-associated (DAM) state in controls.
This editing strategy worked in Cas9 knock-ins, but neither mice nor people express this bacterial gene. Hence the scientists packaged a slimmed-down version of the endonuclease, called SA-Cas9, into the AAV vector along with the APP-targeted guide RNA. Infecting APP-KI mice with that similarly reduced amyloidosis, microgliosis, and lysosomal dysfunction. Ongoing work in Roy’s lab seeks to optimize the guide RNAs, Cas9 endonuclease, and AAV vectors for use in people.
Martin Ingelsson of the University of Toronto pointed out that the gene editing was initiated slightly before plaques start to accumulate, and the mice were sacrificed slightly before they would have had saturated plaque levels in their brains. “To get closer to a ‘real-world clinical scenario,’ it would be relevant to also treat older mice for a more limited period,” Ingelsson wrote (comment below).
While not in the manuscript, Roy told Alzforum that these experiments—in 6-month-old mice—have recently been done. CRISPR editing did not eliminate existing plaques, but it did prevent more from forming, he said. He said the Arctic mutation in these knock-ins yields Aβ plaques that are particularly sticky and stubborn, so their persistence even after APP editing did not surprise him.
Who could be candidates for this gene therapy? Roy thinks it could work for people who have one of the hundreds of known ADAD mutations in APP, presenilin 1, or PS2. To test this idea, he is comparing how well the editing works across human isogenic iPSC-derived neurons from different ADAD mutations.
Eric McDade of Washington University in St. Louis thinks this cell work is critical. “If the outcomes are similar, this could overcome a significant problem of having to target each of the ADAD mutations individually,” he wrote. McDade, a lead investigator in DIAN, was excited to see research move a step closer to use in humans. “This is an approach that many individuals with ADAD mutations have expressed a high level of enthusiasm for,” he wrote (comment below).
Roy said that people with ADAD mutations make ideal candidates for initial human studies. In the future, he envisions using APP editing in people with sporadic AD as well, perhaps in combination with Aβ-targeted antibodies. “After removal of existing amyloid, this one-time gene editing therapy could turn off the tap,” he said.
Kristine Freude of the University of Copenhagen thinks the gene editing elegantly thwarted Aβ-related pathology in mice. “However, translating these findings to humans poses challenges, including delivery methods and the presence of tau pathology,” she wrote. “Investigating the impact of CRISPR/Cas9-mediated APP modifications on tau pathology in AD would be highly relevant.”
Roy’s AAV- and CRISPR-based approach comes at a time when both technologies are making strides in clinical studies. In a first, the FDA approved a CRISPR therapy for sickle-cell anemia last December after approving the AAV-based spinal muscular atrophy therapy Zolgemsa back in 2019 (FDA press release; Nov 2019 news). Other AAV-based biologics are in preclinical and early clinical studies for Gaucher’s, frontotemporal dementia, and other neurodegenerative diseases (Jun 2024 news). Roy thinks clinical studies of his approach are a realistic goal, and has founded the startup CRISPRAlz.—Jessica Shugart
References
News Citations
- Give BACE Inhibitors a Second Chance?
- Moving Forward: RNA-Targeted Attempts at Taking Down Tau, APP
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
- Too Basic: APP β-CTF's YENTPY Motif Binds Proton Pump, Thwarts Lysosomes
- Time to Try Again: Gene-Based Therapy for Neurodegeneration
- All About Exposure: How to Get Enough Progranulin into the Brain?
Research Models Citations
Paper Citations
- Sun J, Carlson-Stevermer J, Das U, Shen M, Delenclos M, Snead AM, Koo SY, Wang L, Qiao D, Loi J, Petersen AJ, Stockton M, Bhattacharyya A, Jones MV, Zhao X, McLean PJ, Sproul AA, Saha K, Roy S. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat Commun. 2019 Jan 3;10(1):53. PubMed.
Other Citations
External Citations
Further Reading
Primary Papers
- Aulston BD, Gimse K, Bazick HO, Kramar EA, Pizzo DP, Parra-Rivas LA, Sun J, Branes-Guerrero K, Checka N, Bagheri N, Satyadev N, Carlson-Stevermer J, Saito T, Saido TC, Audhya A, Wood MA, Zylka MJ, Saha K, Roy S. Long term rescue of Alzheimer's deficits in vivo by one-time gene-editing of App C-terminus. bioRxiv. 2024 Jun 9; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Northwestern University, Feinberg School of Medicine
The authors, in this pre-peer-review manuscript, discuss recent in vivo results of CRISPR-Cas9 editing in the context of Alzheimer's disease. A universal one-and-done gene therapy for AD, which the authors have set as their long-term goal, is alluring, especially in a field that has struggled for decades to find a suitable therapy that will stop the disease process in its tracks.
Their approach, which was previously described in cell culture experiments (Sun et al., 2019), hinges on the spatially separated processing of amyloid precursor protein (APP) by α-secretase and β-secretase, the latter occurring predominantly in the endosomes of the cell where BACE1 (the main β-secretase) is most active due to optimal pH conditions. As the β-secretase pathway is amyloidogenic, producing the toxic Aβ that is a hallmark of AD, the authors' objective was to attenuate APP processing down this pathway by deleting the YENPTY motif at the APP C-terminus, since this motif has been shown to be critical for APP trafficking to endosomes. This approach has the added benefit of increasing APP cleavage by α-secretase, thereby increasing soluble APP-α, which has been reported to have its own neuroprotective benefits.
In this current report, the authors have moved from cell culture to APP knock-in mice to study the feasibility and safety of this CRISPR editing in vivo. Their results show that the approach does shift APP processing away from the amyloidogenic pathway, and into the α-secretase pathway. Not only are Aβ levels lower, but so is lysosomal pathology, astrocytosis, and microglial activation--after just a single dose. Hippocampal LTP improved, and behavioral tests showed improvements in memory in the mice treated with this approach. The authors also generated mice where APP was edited in the germline to evaluate safety, with promising results.
The findings presented are exciting, but a lot more work needs to be done. Certain experiments in this report had very small sample size (two or three mice per group). More mice are needed to instill confidence in these initial results. There are also challenges to gene editing when it comes to delivery into human brains, so these will need to be overcome if the work continues to show promise in preclinical settings. It’s important to also understand that such an approach in people would likely only be beneficial if treatment commenced in early symptomatic or pre-symptomatic stages of AD, before the disease has gotten out of hand. These are the same considerations that we currently have with amyloid antibody therapies—we simply must treat earlier. I’m looking forward to seeing more data from this group in the future!
References:
Sun J, Carlson-Stevermer J, Das U, Shen M, Delenclos M, Snead AM, Koo SY, Wang L, Qiao D, Loi J, Petersen AJ, Stockton M, Bhattacharyya A, Jones MV, Zhao X, McLean PJ, Sproul AA, Saha K, Roy S. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat Commun. 2019 Jan 3;10(1):53. PubMed.
University of Toronto
This yet-to-be-peer-reviewed manuscript by Aulston et al. describes an elegant study in which the authors have investigated whether CRISPR/Cas9-based non-homologous end-joining (NHEJ) can be used in vivo to target the last piece of exon 18 in APP and thereby remove the YENPTY motif at the APP C-terminus. This motif has been shown to be critical in steering APP to endosomes that have β-secretase activity, and by eliminating it more APP should be processed by the non-amyloidogenic pathway. The study follows up on previous findings by the authors, in which they have showed, both ex vivo in cells and in vivo in mice (intracranial only), that their approach does have its intended effects on APP processing. In the current study, they demonstrate that the treatment, when administered peripherally (via retro-orbital injections), can also lead to desired neuropathological, biochemical, electrophysiological, and behavioral effects on APPNL-G-F mice. If the treatment is initiated at 1.5 months they can see an amelioration of pathology, whereas if they intervene already at the germline stage they can virtually abolish plaque development.
The authors suggest that their proposed gene-editing strategy should enable a universal and efficient one-time treatment to prevent brain pathology in AD. However, whereas the suppression of C-terminal APP is dramatic, the effects on plaque load and Aβ levels are not as robust, indicating that the residual β-secretase cleavage is sufficient for progression of pathology if treatment is not initiated already at the germline stage. The clinical effects (as measured by improved novel object recognition) are even less pronounced. Moreover, compared to the outcome of the preclinical and clinical studies of anti-Aβ immunotherapies it is (based on this study) not clear to me that gene-therapy-mediated disruption of the APP C-terminus would be more efficient, at least not if treatment is started on individuals who are already symptomatic. It is of course hard to make fair comparisons, as the authors, in the efficacy part of the study, initiated their editing slightly before plaques start to accumulate and sacrificed the mice slightly before plaque load can be expected to have saturated their brains. To get closer to a “real-world clinical scenario,” it would be relevant to also treat older mice for a more limited period. This would more closely mimic the clinical situation.
Either way, NHEJ-based gene editing is an exciting avenue for treating diseases where it can be assumed toxic gain of function is at play. As for AD, the APP-targeting approaches mainly offer possibilities to prevent generation and buildup of pathogenic Aβ species and would therefore be most efficient in the preventive setting. The work by Subhojit Roy and colleagues represents one of several strategies that, together, provide a framework for potential future Aβ-lowering gene-editing therapies.
Washington University
Subhojit Roy and his team report exciting transgenic-mouse work, targeting the amyloid precursor protein (APP) with CRISPR-Cas9/AAV. This follows nicely on Dr. Roy’s cell-based work and provides promising, preliminary evidence for using this approach to target the common pathway for nearly all autosomal dominantly inherited Alzheimer’s disease (ADAD) mutations. The technical aspects are far outside my area of expertise, but I have a few observations from this preprint.
The targeting of the C-terminal appears to provide a significant upstream effect on the β/γ-secretase processing of APP. The broad attenuation of endosomal and inflammatory/immune pathways suggests that this approach could have widespread effects on the pathological cascade of AD, and the lowering of endosomal markers is an important pharmacodynamic validation of this approach. In the current study, the triple knock-in represents three different APP mutations. Although each of these likely has different mechanisms for causing early onset AD, it is a limitation. Future work that includes PSEN1 and PSEN2 mutations will be helpful to demonstrate that this approach could have broader implications for ADAD. If the outcomes are similar, this could overcome a significant problem of having to target each of the identified ADAD mutations.
Of note, there remained substantial amyloid plaque and microglial, astrocytic, and endosomal markers in the CAS9/AAV mice, even after the initial injections were given prior to significant plaque accumulation, but this seemed to be driven by homozygous mutations. In heterozygous mutations, more consistent with the majority of mutations in humans, early CRISPR injection nearly eliminated plaques, suggesting a possibility for early interventions in ADAD mutation carriers. Likewise, targeting heterozygotes seemed to have similar, broad effects on lysosomal, inflammatory pathways, with the exception of an increase in astrocyte-related markers that might be a concerning sign of a reaction to the AAV. How consistent this will be, and how long it may last, will be important considerations prior to considering clinical trials in humans. The reversibility of this inflammation, or any other potential off-target effects, remains a major concern for CNS-related strategies.
Lastly, it would be helpful to see the effects of targeting different aspects of the APP gene. Although this study suggests a high-level of specificity for the C-terminal exon, and a low level of off-target effects, it would be important to know what happens if other areas are down-regulated.
It is exciting to see this type of work moving one step closer to potential use in humans. I know that this is an approach that many individuals with ADAD mutations have expressed a high level of enthusiasm for. However, it will be very important for the scientific community to communicate how close (or far) we are to getting this into clinical trials, so that our research partners understand what remains to be done before starting these types of trials.
University Copenhagen
Aulston et al. provided compelling evidence that targeting the C-terminal coding region of amyloid precursor protein (APP) reduces its processing by β-secretase (BACE1). This reduction in BACE1 activity attenuates the amyloidogenic pathway and decreases the production of plaque-forming Aβ peptides. The study employed a CRISPR/Cas9 approach to target the sequence encoding the YENPTY motif, which interacts with BACE1. As the nucleotides encoding this motif are in the most 3' exon, the authors facilitated non-homologous end joining (NHEJ) using their CRISPR/Cas9 technique. This led to a truncated version of APP with reduced BACE1 interaction capacity. In vivo mouse models showed that not only BACE1 activity but also plaque load and neuroinflammation were reduced.
These intriguing results raise the question of whether the observed effects on glial cells are direct, or a consequence of reduced Aβ40 and Aβ42 production. Overall, this elegant study successfully reversed Alzheimer’s disease pathology related to Aβ in mouse models. However, translating these findings to humans poses challenges, including delivery methods and the presence of tau pathology. Investigating the impact of CRISPR/Cas9-mediated APP modifications on tau pathology in Alzheimer's disease would be highly relevant.
Icahn School of Medicine at Mount Sinai
In 1993, we investigated whether APP is present in clathrin-coated vesicles (CCVs) (Nordstedt et al., 1993). CCVs are responsible for the trafficking of many proteins to the endosomal compartment, including the transport of plasma-membrane receptors to early endosomes, and of proteins destined for lysosomes from the trans-Golgi network to late endosomes and pre-lysosomes. While little is known about the mechanism(s) whereby integral membrane proteins are targeted to the population of CCVs that exit the trans-Golgi network, specific signals have been identified that are responsible for targeting cell-surface proteins to the CCVs that bud off the plasma membrane. Both the influenza virus hemagglutinin molecule and the cation-independent mannose 6-phosphate receptor are efficiently endocytosed but only if a tyrosine residue is present at specific locations in their cytoplasmic domains. Endocytosis of the low-density lipoprotein receptor requires an asparagine-proline-X- tyrosine (NPXY, where X represents any amino acid) motif in its cytoplasmic domain, perhaps enabling its interaction with the assembly and adaptor proteins that are believed to mediate the interactions between internalized proteins and the clathrin cage of the CCV. APP is among the numerous cell-surface proteins possessing an NPXY motif in their cytoplasmic domains, suggesting that it too might be targeted to, and internalized via, clathrin-coated pits. The identification of APP in CCVs provided direct evidence for the trafficking of APP to the endosomal/lysosomal system. In our 1993 report, we described the purification of CCVs from PC12 cells, using a combination of protocols developed for the isolation of CCVs from mammalian organs. The CCV preparation was characterized by SDS-polyacrylamide gel electrophoresis, electron microscopy, and immunoblot analysis using as a marker the transferrin receptor, an internalized protein known to be present in CCVs. We showed that full-length mature (fully posttranslationally modified) APP and the α and β carboxyl-terminal fragments resulting from APP secretory cleavage are enriched in CCVs.
Aulston et al. built on this in their current paper. They used CRISPR-Cas9 to edit the last exon of mouse amyloid precursor protein (App) gene. Their approach eliminated the endocytic YENPTY motif at the APP C-terminus, while preserving the N-terminus and compensatory APP-homologues (APLP1, APLP2). The CRISPR reaction favorably altered events along the Aβ peptide generating pathway by reducing toxic APP-β-cleavage fragments, while also upregulating neuroprotective APP α-cleavage products. AAV-driven editing prevented the neuropathologic, electrophysiologic, and behavioral features of a popular Alzheimer’s knock-in mouse model. Effects were persistent, and no abnormalities were seen in wild-type mice, even after germline App editing, thereby underlining the overall efficacy and safety of the editing reaction. Pathologic alterations in the glial transcriptome of App-KI mice, as seen by single nuclei RNA-sequencing were also normalized by App C-terminus editing.
Aulston et al.’s strategy takes advantage of innate transcriptional rules that render terminal exons insensitive to nonsense decay, and the upstream manipulation is expected to be effective for all dominant and sporadic forms of Alzheimer’s disease. Even more surprisingly, these studies could form the basis for a one-time disease-modifying treatment for Alzheimer’s disease.
References:
Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993 Jan 5;268(1):608-12. PubMed.
University of California, San Diego
Admitting a conflict of interest in commenting on this research (Subhojit's office is two doors down from mine), I find this work to be very promising. The idea of editing even normal APP to remove potential *future* toxic N-terminal species makes it relevant to treatment not just of autosomal-dominant mutant causes of AD, but sporadic AD also.
New generation AAV vectors are under development in very active academic, biotech, and pharma research programs. Directed evolution is producing novel synthetic AAV serotypes capable of broadly infecting the CNS in primates while de-targeting peripheral infection, especially in the liver. Thus, future therapy could be accomplished with simple IV vector infusion, albeit at considerable cost, to a large population.
I, too, share the opinion that starting early—possibly before amyloid accumulation—may be key in having a clinical impact with this type of amyloid-targeting gene therapy approach.
Meanwhile, we are continuing to enroll patients in a Phase 1 study of AAV2-BDNF gene therapy in early AD and MCI. Our aim is to protect neural circuits through a different mechanism: neurotrophic factors. BDNF prevents cell death, activates neuronal function and rebuilds synapses in animal models of AD (Nagahara et al., 2009). We have treated three patients to date, and there have been no safety concerns. We will treat nine additional patients in this Phase 1 trial.
In the future, one might combine amyloid-targeting gene therapy and neurotrophic gene delivery. This would have the potential to both mitigate an AD risk factor (APP) and address ongoing neurodegeneration from other mechanisms (Tau, aging) to prevent cell death and boost remaining neurons.
References:
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009 Mar;15(3):331-7. PubMed.
Make a Comment
To make a comment you must login or register.