Tweaked, Aβ-Antibodies Cross Blood-Brain ‘Border’ (Bye-Bye, Barrier?)
Quick Links
Far from a static wall, the so-called “blood-brain barrier” is a dynamic, bustling interface that allows select groups of molecules to pass in and out of the brain. Which proteins and nutrients can cross varies according to the needs of the cells on either side. While probing the nuances of this “border,” scientists are honing techniques for transporting therapeutics into every nook and cranny of the brain’s parenchyma, while circumnavigating the brain’s amyloid-laden arteries. Case in point, in a preprint posted on bioRxiv on August 14, Joy Zuchero and colleagues at Denali Therapeutics in South San Francisco described their latest overhaul of an antibody transport vehicle.
- Halving effector function of a transferrin receptor-targeted antibody transport vehicle spares red blood cells.
- This ATV still targets Aβ in the brain.
- Compared to standard antibodies, the ATV distributes broadly across the mouse brain. It doesn’t cause ARIA.
- Scientists propose changing the term blood-brain barrier to blood-brain “border.”
ATVs are antibodies sporting a transferrin receptor (TfR) binding site within one of their two effector domains. In mouse models, the authors found that halving the effector function of this construct mitigated depletion of TfR-expressing red blood cells in the periphery, but maintained the antibody’s ability to vanquish its target in the brain—in this case, Aβ plaques. Compared to a regular anti-Aβ antibody, the ATV version was better distributed throughout the brain and provoked less ARIA in perivascular regions.
“Overall these are very encouraging results that could eventually lead to new and safer treatment paradigms for AD,” commented Stephen Salloway of Brown University in Providence, Rhode Island.
The preprint comes on the heels of a review penned by Denali’s Robert Thorne along with a cadre of academic scientists. Together, they offer a reconceptualization of the BBB, proposing that the last “B” should stand for “border” instead of “barrier.” Not unlike a border that allows for regulated exchange of people and goods tailored to a country’s specific needs and relationships, the interfaces between the blood and brain are also selective and adaptive, they argue (see next story).
A scant proportion of large molecules make it from the blood into the brain. For therapeutic monoclonal antibodies, that’s at most 0.5 percent of what’s in the periphery. Those that do get in tend to cross at the interface between the blood and CSF, rather than via the capillaries crisscrossing the parenchyma. Many of the border crossers may then get waylaid within CSF-adjacent spaces, holding up distribution throughout the parenchyma and arrival at their intended targets, such as Aβ plaques.
To get around this problem, scientists at Denali and at other companies take advantage of the transferrin receptor. The TfR resides on the surface of endothelial cells lining the brain vasculature, with the highest expression in the smallest vessels. Called ATV, Denali’s construct features a TfR binding domain integrated into one of the Fc domains of an antibody (May 2020 news). Preclinical studies are ongoing for ATVs against BACE1, TREM2, Aβ, and other targets.
Another TfR-based approach is Roche’s Brain Shuttle, in which a TfR-specific Fab fragment is attached to the end of one of the two Fc regions of a therapeutic antibody. Trontinemab—essentially gantenerumab equipped with a Brain Shuttle—is in Phase 1/2. It entered the brain more readily than its predecessor while provoking hardly any amyloid-related imaging abnormalities (Mar 2024 conference news). The vascular changes underlying ARIA are the Achilles heel of Aβ-targeted therapies, and scientists are searching for ways to avoid them (Aug 2024 conference news).
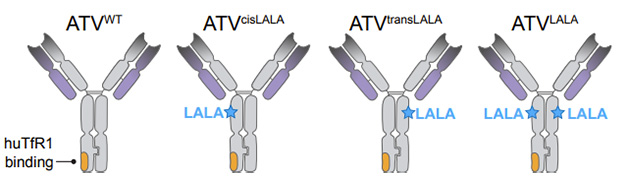
LALA to the Rescue. Effector-quashing LALA mutations were introduced into either or both Fc domains of ATV constructs. Only when LALA resided on the same Fc domain (ATVcisLALA or ATVLALA) as the TfR binding site were red blood cells spared. [Courtesy of Pizzo et al., bioRxiv, 2024.]
One downside of using the TfR is that reticulocytes—the predecessors of red blood cells—also express it. Indeed, trontinemab and early versions of Denali’s ATVs target these cells. This can lead to anemia.
Previously, Denali scientists reported that introduction of two mutations—L234A/L235A, aka LALA—into the Fc effector domains stifled this anti-RBC activity. Trouble is, these mutations also compromised the antibodies’ effector function against their targets. For the current study, first author Michelle Pizzo and colleagues struck a compromise, inserting the LALA mutations into only one of the ATV’s Fc domains. In short, when they introduced these effector-dampening mutations into the same Fc domain housing the TfR binding region—cisLALA—it substantially doused the antibody’s effector activity directed against TfR-expressing cells. When they introduced cisLALA into an anti-Aβ ATV and injected that into TfRmu/hu mice that express human TfR, it spared red blood cells. In contrast, RBCs plummeted in mice injected with ATV-Aβ constructs that lacked the LALA mutations, or only carried them on the opposing (trans) Fc region.
Roche scientists led by Per-Ola Freskgård, now at BioArctic, previously reported that the attachment of their TfR-targeted Fab fragment to the end of their antibody’s Fc domain blocked the domain’s effector function when the construct was bound to TfR (Jan 2018 news). Therefore, they concluded that no effector-dampening mutations were needed.
How would this configuration compare to cisLALA in terms of sparing red blood cells in mice? To find out, Pizzo and colleagues compared them head-to-head. They attached a TfR-targeted Fab fragment to the Fc region of their fully functional anti-Aβ antibody. When injected into mice, it triggered a reduction in reticulocytes, while the ATVcisLALA antibody did not. The results suggested that their new approach to building ATVs blunts the loss of erythrocytes, the authors concluded.
Despite sparing reticulocytes, the cisLALA mutations did not undermine the antibody’s ability to attack the target of its Fab arms, i.e., Aβ plaques. The scientists injected 5xFAD mice every three days for 12 days with a high dose of different antibody constructs, achieving a similar brain exposure for each. They found that the ATVcisLALA-Aβ antibody recruited about as many microglia to Aβ plaques as did the unmodified anti-Aβ antibody, whereas the effectorless ATV-LALA-Aβ antibody summoned fewer microglia. Both the unmodified anti-Aβ and ATVcisLALA-Aβ similarly reduced the area of small Aβ plaques, though neither put a dent in large plaques during this short treatment.
Freskgård noted that according to in vitro experiments described in the manuscript, ATVCisLALA did slightly dampen the antibody’s cytotoxicity against Fab-bound targets. He wondered if this might lower potency in removing Aβ plaques. “It would be great to see more detailed in vivo potency data addressing this aspect,” Freskgård wrote to Alzforum.
The scientists next compared the brain distribution of their ATVs to that of standard antibodies, and assessed how distribution affected the treated brain’s vascular woes. They found that after a single dose, the concentration of ATVCisLALA-Aβ in total brain lysates topped that of a regular anti-Aβ antibody by five- to eightfold. Crucially, the whereabouts of the ATV and unmodified antibodies were strikingly different. While anti-Aβ antibodies milled about in CSF-adjacent, perivascular spaces, ATVCisLALA-Aβ pervaded the parenchyma of an entire sagittal brain section, including deep brain regions (image below).

Down to Distribution. Unmodified anti-Aβ antibodies (top row) congregated along connected regions in the choroid plexus (middle), while ATVCisLALA-Aβ antibodies (bottom row) fanned out across the cortical surface and associated with small structures (right).
Deploying tissue clearing followed by three-dimensional imaging, the scientists saw that while unmodified, anti-Aβ antibodies seemed stuck along what appeared to be major surface arteries such as those coursing through the leptomeninges, ATVCisLALA-Aβ huddled with small dots distributed throughout the parenchyma. Separate staining of the other half of each brain suggested that these dots were parenchymal amyloid plaques, while the larger, connected swaths bound by the anti-Aβ antibodies were likely vascular amyloid, akin to cerebral amyloid angiopathy (CAA).
Would this even distribution of ATVCisLALA-Aβ sidestep ARIA? The scientists strapped their ATV construct to a notorious ARIA instigator: a mouse version of 3D6, aka bapineuzumab. In 5xFAD mice, this antibody reportedly makes a good ARIA model (Mar 2024 conference news). Indeed, as per mouse MRI, the unmodified 3D6 antibody provoked ARIA-like events in all 10 5xFAD mice used in this study, while an equal dose of ATVCisLALA-3D6 triggered a single ARIA-like lesion in one mouse, despite its higher penetration into the brain. When matched with unmodified 3D6 for brain exposure, ATVCisLALA-3D6 provoked nary any ARIA. Neither did ATV-3D6, which retained full effector function.
The MRI findings jibed with what the researchers found via histopathology, namely, that ushering 3D6 into the brain via ATV neutralized its inflammatory threat against the cerebrovasculature, while unmodified 3D6 antibodies wreaked havoc.
To Zuchero and co-author Thorne, the data suggest that sans ATV, antibodies trickle into the brain via a tortuous, indirect route. They first pass from blood into CSF, then work their way across the brain’s surface and into the perivascular spaces around large-caliber leptomeningeal vessels. This is where vascular amyloid accumulates—ground zero for ARIA. “By contrast, our ATV:Aβ molecule distributes to the brain mostly by transport across the microvessels which highly express the transferrin receptor, essentially bypassing the problematic part of the vessel where interaction with vascular amyloid can lead to ARIA,” Zuchero and Thorne wrote to Alzforum.
Costantino Iadecola of New York’s Weill Cornell Medical College called the findings promising and timely. Risk of ARIA besets the approved anti-Aβ antibodies (Aug 2024 conference news).
Iadecola said that Pizzo’s findings jibe with one hypothesis about what causes ARIA, i.e., that conventional, intravenous antibodies get marooned in perivascular spaces, where CAA happens. This convergence instigates a pro-inflammatory storm that destabilizes vessel walls, leading to the swelling and hemorrhage that presents as ARIA. Iadecola favors a variation of this, whereby the therapeutic antibody itself exacerbates Aβ accumulation in the perivascular space, which riles macrophages there. Iadecola recently reported that the resulting vascular damage is worse when these border-associated macrophages express ApoE4 (Iadecola et al., 2024). Given that, he wondered how Pizzo et al.’s experiments would turn out in ApoE4 mice.
Another view is that immune complexes between Aβ and antibodies transform microglia into a maladaptive state, causing ARIA, Iadecola noted (Aug 2024 conference news). He thinks it’s possible that all three mechanisms play a role. “With something as complex as the brain vasculature, it’s never just one thing,” he told Alzforum.
Given this complexity, Thorne and other scientists recently made the case to view the BBB more like an actively managed, dynamic border between countries, rather than a static wall. For more on that, check out our next story (Oct 2024 news).—Jessica Shugart
References
News Citations
- Reconceptualizing the BBB: Is It Time to Swap ‘Barrier’ for ‘Border'?
- Molecular Transport Vehicle Shuttles Therapies into Brain
- Fast Plaque Clearance with Little ARIA? So Teases Trontinemab at AD/PD 2024
- Two New Deaths on Leqembi Highlight Need to Better Manage ARIA
- Antibody Shuttle Rouses Anti-Aβ Response in Brain without Waking the Periphery
- Mouse Models and Markers for Cerebral Amyloid Angiopathy, ARIA
- Implicated in ARIA: Perivascular Macrophages and Microglia
Therapeutics Citations
Paper Citations
- Iadecola C, Anfray A, Schaeffer S, Hattori Y, Santisteban M, Casey N, Wang G, Strickland M, Zhou P, Holtzman D, Anrather J, Park L. Cell autonomous role of border associated macrophages in ApoE4 neurovascular dysfunction and susceptibility to white matter injury. Res Sq. 2023 Aug 4; PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Pizzo ME, Plowey ED, Khoury N, Kwan W, Abettan J, DeVos SL, Discenza CB, Earr T, Joy D, Lye-Barthel M, Roche E, Chan D, Dugas JC, Gadkar K, Meisner R, Sebalusky J, SilvaAmaral AC, Becerra I, Chau R, Chow J, Clemens AJ, Dennis MS, Duque J, Fusaro L, Getz JA, Kariolis MS, Kim DJ, Leung AW, Nguyen HN, Thomsen ER, Sanchez PE, Shan L, Silverman AP, Solanoy H, Tong R, Calvert ME, Watts RJ, Thorne RG, Weinreb PH, Walsh DM, Lewcock JW, Bussiere T, Zuchero YJ. Engineering anti-amyloid antibodies with transferrin receptor targeting improves safety and brain biodistribution. 2024 Jul 26 10.1101/2024.07.26.604664 (version 1) bioRxiv.
Annotate
To make an annotation you must Login or Register.
Comments
Brown University
Research groups at Denali and Roche have demonstrated encouraging preliminary results in amyloid reduction with lower ARIA rates using plaque-targeting antibodies bound to an antibody transport vehicle targeting the transferrin receptor. Both groups propose broad brain parenchymal antibody distribution with lower binding to vascular Aβ, a key mechanism suspected of underlying the production of inflammation and ARIA. The Denali team used an animal model of amyloid deposition but still needs to conduct this work in AD patients. Their molecular profile seems to mitigate some of the transferrin-related hematological effects. Early data from the Roche trontinemab Phase 1b program shows robust reduction of plaque burden with limited ARIA after just a few doses. This will need to be demonstrated in larger samples with sufficient duration to test for a clinical benefit. Overall these are very encouraging results that could eventually lead to new and safer treatment paradigms for AD.
Make a Comment
To make a comment you must login or register.