Without SORL1, Endosomes Swell in Neurons but not Microglia
Quick Links
Scientists have linked loss-of-function mutations in SORL1 to overproduction of Aβ and to early onset Alzheimer’s disease. Now, in the June 2 Cell Reports, researchers led by Jessica Young at the University of Washington, Seattle, report that knocking out the gene for this sorting receptor in human neurons causes early endosomes to enlarge. At the same time, trafficking of the Aβ precursor protein (APP), and likely other cargo, slowed down. This happened only in neurons, not microglia. The SORL1 knockout recapitulates one of the earliest cytopathological hallmarks of AD, Young told Alzforum.
Perhaps counterintuitively, a β-secretase inhibitor didn’t keep the organelles from swelling. “We show that removal of SORL1 causes enlargement of endosomes independently of APP processing,” said Young. She presented most of this data at AAIC last year (Sep 2019 conference news).
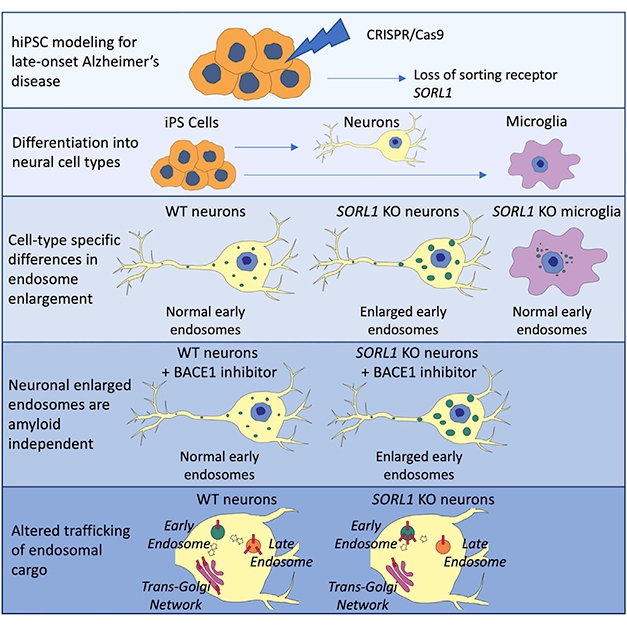
KO Model. CRISPR knocks SORL1 out of pluripotent stem cells, allowing scientists to derive SORL1-free neurons and microglia. Only the former showed signs of endosomal stress and APP trafficking deficits. [Courtesy of Knupp et al., Cell Reports.]
Also last year, researchers led by Marc Tessier-Lavigne at Stanford University, California, reported that endosomes become enlarged in neurons carrying familial AD mutations (Aug 2019 news). His and Young’s findings point to two different sources of endosomal dysfunction in AD—mutations in APP or presenilin, and loss of function of the SORL1 receptor. “This suggests a pivotal, early, upstream mechanism in the disease,” Young said. Scientists had first reported endocytic problems in AD 20 years ago (Cataldo et al., 2000).
To mimic loss of SORL1 function, co-first authors Allison Knupp and Swati Mishra used CRISPR to knock the gene out of pluripotent stem cells. They induced these from fibroblasts taken from a healthy man who carried one ApoE4 allele. Knupp then used SORL1 knockout and isogenic wild-type cells to derive neurons and microglia.
In the SORL1-knockout neurons, two-photon microscopy detected enlarged puncta containing the early endosome markers EEA1 and Rab 5 (see image below). Trafficking of APP, which normally moves through these early endosomes back to the cell membrane or trans-Golgi network (TGN), seemed perturbed. Again using two-photon imaging, the researchers found more APP in EAA1 compartments and less in the TGN than in wild-type neurons.
Adding the discontinued BACE inhibitor LY2886721 did not reverse these endosome changes, even though it reduced production of sAPPβ, APP β-C-terminal fragments, and Aβ peptide levels. “The findings suggest that it would be short-sighted to think about SORL1 solely in terms of protecting against Aβ or amyloid production,” said Young. “It has other functions that keep neurons healthy.” While the SORL1 knockout neurons seemed as viable as wild-type, Young thinks that over time, trafficking defects would starve neurons of growth factors or receptors necessary for function and survival.
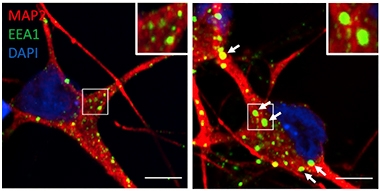
Swollen Endosomes. Neurons (red) derived from SORL1-negative iPSCs (bottom) contain larger and more numerous early endosomes (green) than do wild-type neurons (top). [Courtesy of Knupp et al., Cell Reports.]
Curiously, microglia seemed unperturbed by the loss of SORL1, even though they express as much of the sorting receptor as do neurons. Young is not sure why the microglia get off so easily. “This likely reflects the intricacies of different cells in the brain,” she said. She acknowledged that these are cultured microglia and might behave differently if they became activated. “That is something critical to think about,” she said.
Young plans to use CRISPR to knock in AD-related loss-of-function variants in the SORL1 gene to see if they behave similarly to the knockouts.—Tom Fagan
References
News Citations
- Can Induced Neurons Identify Early Signs of Neurodegeneration?
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
Therapeutics Citations
Paper Citations
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
Further Reading
Primary Papers
- Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, Young JE. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020 Jun 2;31(9):107719. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Aarhus University
The work from Jessica Young’s group is elegant and adds further to the wealth of evidence that place SORL1 as a central player in Alzheimer’s disease.
They find that when neurons derived from iPSCs are genetically modified to inactivate SORL1, they show trafficking and processing defects of APP, confirming previous findings by others and us, and that SORLA determines endosomal trafficking of APP. However, the current study now links yet another AD pathology to SORL1 activity, as these neurons display enlarged endosomes, similar to the cytopathology hallmark for neurons from AD brains. Interestingly, endosomal enlargement following SORL1 inactivation was only found in neurons but not in microglia. This finding is in line with our paper from last year, which reported SORL1 silencing leading to enlarged (and dysfunctional) lysosomes in only some cells (Pietilä et al., 2019).
SORL1 is (misleadingly?) often referred to as a ”microglia gene.” This is likely due to high transcript levels seen in activated microglia in AD brains. However, it is well known that for many genes there is little correlation between transcript and translated protein levels. And to date there is no proof that SORLA has any functional role in microglia. This paper by the Young group does not change that, although this is now the first time that SORLA protein expression was demonstrated for human microglia.
Future studies should aim to determine if SORLA has any function in this cell type. However, all evidence so far points to neuronal functions of SORLA, and the current paper provides further support for how SORL1 inactivation leads to neuronal defects, which in my view is critical for understanding the link between SORL1 and Alzheimer’s disease.
References:
Pietilä M, Sahgal P, Peuhu E, Jäntti NZ, Paatero I, Närvä E, Al-Akhrass H, Lilja J, Georgiadou M, Andersen OM, Padzik A, Sihto H, Joensuu H, Blomqvist M, Saarinen I, Boström PJ, Taimen P, Ivaska J. SORLA regulates endosomal trafficking and oncogenic fitness of HER2. Nat Commun. 2019 May 28;10(1):2340. PubMed.
Make a Comment
To make a comment you must login or register.