In Familial AD, Amyloid-PET Varies by Mutation Type; CSF Aβ42 Doesn't
Quick Links
Bad as they are, the hundreds of mutations in the APP, PSEN1, and PSEN2 genes that cause autosomal-dominant AD are not all the same in severity. Many bring on symptoms in a carrier's 40s and 50s, but their range spans onset in the 20s to the 70s. A new study, led by Jasmeer Chhatwal of Brigham and Women’s Hospital in Boston and published in the February issue of Lancet Neurology, suggests that different mutations trigger different patterns of amyloid accumulation with respect to the carrier's estimated timing of symptom onset. In other words, amyloid deposition as measured by PET does not track consistently with the clinical course of the disease. The situation was different for CSF Aβ42 levels, which did not differ much by mutation type.
- In autosomal-dominant AD, mutation type influenced amyloid-PET pegged to estimated years before symptom onset.
- Some mutations came with more striatal amyloid; some with cortical.
- CSF Aβ42 did not vary by mutation; neither did clinical decline.
To Niklas Mattsson-Carlgren of Lund University in Sweden, the study indicates that amyloid PET does not provide consistent information about AD stage among carriers of autosomal-dominant AD mutations.
“I agree with the authors that this supports the hypothesis that amyloid PET and CSF β-amyloid are not completely interchangeable and likely provide complementary information in ADAD, which has also been shown before in sporadic AD,” he wrote.
“The heterogeneity has very important implications for anti-amyloid trials in ADAD when using Aβ-PET to determine target engagement, and when correlating reductions in Aβ-PET signal with clinical outcome measures,” added Lund’s Oskar Hansson.
Despite being a hallmark of AD, amyloid’s relationship with the clinical progression of disease has always been tenuous, particularly for people with sporadic, late-onset forms. Myriad variables—genetic, environmental, and lifestyle—can tweak the relationship between plaques and cognitive decline, as do other brain pathologies that become more prevalent with age. People with autosomal-dominant AD, who generally develop AD symptoms earlier in life and at a similar age within each affected family, have fewer of these confounders.
“These unfortunate mutations offer us a rare bit of terra firma,” Chhatwal told Alzforum. “They allow us to ask how faithfully our biomarkers reflect disease progression.” That was the focus of the current study, which made use of cross-sectional and longitudinal measurements of amyloid burden in 340 participants in the dominantly inherited AD network (DIAN), including 206 mutation carriers and 134 unaffected family members.
The researchers started by grouping participants by mutation type in two different ways. The first was based on which protein domain was affected by the underlying mutation in each gene. Each of nine domain-based mutation groups included a minimum of 10 carriers from at least two families. The groups spanned mutations in the transmembrane domains of APP and PSEN2, as well as variants within the cytoplasmic domain and several transmembrane domains of PSEN1. The second grouping method only pertained to the 161 PSEN1 mutation carriers in the cohort. The researchers divided these mutations into two sets: those that landed before, or after, codon 200.

Sort in Search of Patterns. Locations of 62 pathogenic mutations within PSEN1, PSEN2, and APP genes. Mutations were grouped by which protein domain they affect, and by whether they lie before or after codon 200 in PSEN1. [Courtesy of Chhatwal et al., Lancet Neurology, 2022.]
Cross-sectional data from the entire study cohort showed that mutation carriers had lower CSF Aβ42 and a greater amyloid burden as per PiB-PET than did noncarriers, and both biomarkers became more abnormal the closer a participant was to his or her estimated year of onset (EYO). For cognition, scores on the clinical dementia rating scale sum of boxes (CDR-SB) started to rise as participants approached their EYO and then ramped up steadily thereafter. But when the researchers compared these three variables across different mutation groups, they noticed that while CSF Aβ42 and CDR-SB scores changed similarly with respect to EYO, PiB-PET was a different beast. Whether the mutations were grouped by domain or by position, the relationship between amyloid burden and EYO was significantly different between groups (see image below).
For PSEN1, carriers of mutations affecting cytoplasmic domain 4, or transmembrane domains 6 or 8, had less cortical PiB binding in relation to their EYO than did carriers of other mutations. By contrast, PSEN1 transmembrane domain 3 and 5 and PSEN2 transmembrane domain 2 mutation carriers had a higher cortical Aβ burdens relative to their disease stage than did other carriers. People with a pathogenic variant that landed before codon 200 in PSEN1 also had significantly more amyloid relative to their EYO. What’s more, the relationship between PiB-PET and CSF Aβ42 varied significantly by mutation group.
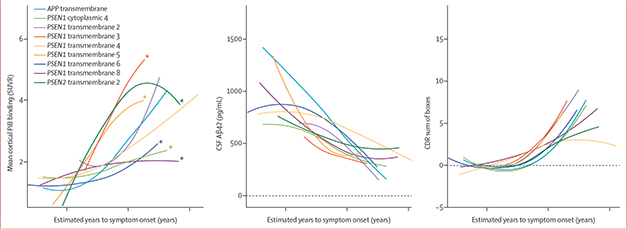
PiB-PET Goes Rogue. Among people with familial AD, the correlation between amyloid burden (left, y-axis) and estimated years from symptom onset (x-axis) varied with the protein domain that was mutated. Neither CSF Aβ42 (middle) nor CDR-SB scores (right) differed by mutation group. [Courtesy of Chhatwal et al., Lancet Neurology, 2022.]
Among 77 participants with PSEN1 mutations who had undergone serial PiB-PET scans, those with mutations before codon 200 accumulated amyloid more quickly. This held only for people who had clinical symptoms. PiB-PET change was similar across both codon categories among people in the asymptomatic stage of AD.
Besides influencing the timing and overall load of amyloid plaques, could mutations dictate where in the brain plaques grow? To address this, the scientists compared carriers’ PiB-PET signals in the cortex with those in the striatum. They found that some mutation groups had a more striatal or cortical-predominant pattern of amyloid deposition, echoing an early report of this curious finding from the dawn days of PiB-PET (Klunk et al., 2007). Specifically, the PSEN1 transmembrane domain 4 and PSEN2 transmembrane 2 mutation groups were cortical-leaning, while the PSEN1 transmembrane 2 and PSEN1 transmembrane domain 8 groupings had a striatal bent.
Finally, the researchers compared the ratio of CSF Aβ42 to Aβ40 across carriers. They found that the ratio declined as the disease timeline progressed. This ratio differed among mutation groups with respect to EYO; however, the differences did not track with those observed in PiB binding, suggesting that differences among mutation groups in the Aβ42/40 ratio do not explain differences seen on the scans.
In all, the findings suggest that for familial AD, amyloid burden as measured by PiB-PET does not consistently track with disease stage, Chhatwal said. Why not? The authors point to differences in how each mutation affects APP processing, which could generate different suites of Aβ peptides churned out by γ-secretase. Longer peptides—Aβ42 and Aβ43—contribute more to the fibrillar Aβ plaques PiB-PET detects than do shorter peptides, hence a shift in a person's Aβ peptide repertoire could increase or decrease the amount of amyloid the scans pick up, said Chhatwal.
David Holtzman of Washington University, St. Louis, had a similar take. He thinks the type of amyloid a person has, and how it binds PET ligands, might be different depending on his or her FAD mutation. “These results, along with prior studies, clearly bring out that detection of amyloid fibrils in the brain by amyloid imaging does not always reflect the location or amount of all the Aβ that is aggregated in the brain, especially when comparing different ADAD mutations,” he wrote to Alzforum (see comment below).
An extreme example of this phenomenon is the Arctic mutation in APP. It leads to aggregation of protofibrillar forms of Aβ that form diffuse, ring-like plaques and are poorly detected by PiB-PET (Basun et al., 2008; Schöll et al., 2012). “The absence of PiB binding seen in the Arctic mutation carriers suggests that the presence of other forms of Aβ, such as oligomers and protofibrils, are important for the pathologic processes leading to clinical AD,” commented Lars Lannfelt of Uppsala University in Sweden, who discovered this mutation.
Another example of this phenomenon is the known absence of a PiB-PET signal in inner cortical layers that do contain aggregated Aβ as seen by other methods, Lannfelt wrote (full comment below).
In sporadic AD, there have been cases who are PiB-PET negative and CSF Aβ42-positive, Holtzman added. At autopsy, they were found to have mostly non-fibrillar Aβ plaques (Cairns et al., 2009).
Hansson noted another variation on this theme, that is, the idea that mutations might differ in the types of plaques they cause. “It would be interesting to determine if regional differences in the ratios between diffuse and dense-core Aβ plaques vary across APP and PSEN mutations, whereby PiB preferentially binds to dense-core plaques, but reduced CSF Aβ42 levels likely associate with both types,” he wrote.
“PiB-PET and Aβ42 or Aβ42/40 are reasonably good surrogate markers for pathogenic aspects of AD,” wrote Lannfelt. “However, they are not pinpointing the real toxic species of Aβ, which most likely are the soluble aggregated species of Aβ, i.e., oligomers and protofibrils, and this might explain the bad correlation to disease stage when using the present methods for measuring Aβ burden.”
Researchers interviewed for this story agree that amyloid-PET should not be used as the sole biomarker of disease progression in AD clinical studies. What about CSF Aβ42? Chhatwal said because there is substantial variability in the relationship between CSF Aβ42 and EYO between individuals, particularly at early stages, CSF Aβ42 alone is not a perfect reflection of disease stage, either. Then how about markers, such as tau? Ongoing studies are currently assessing how tau biomarkers, including tau-PET as well as fluid species of phospho-tau, track with disease stage in the DIAN cohort.—Jessica Shugart
References
Mutation Interactive Images Citations
Mutations Citations
Paper Citations
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, Dekosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007 Jun 6;27(23):6174-84. PubMed.
- Basun H, Bogdanovic N, Ingelsson M, Almkvist O, Näslund J, Axelman K, Bird TD, Nochlin D, Schellenberg GD, Wahlund LO, Lannfelt L. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008 Apr;65(4):499-505. PubMed.
- Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, Almkvist O, Graff C, Nordberg A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012 Jul 17;79(3):229-36. PubMed.
- Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, Reinwald LT, Carter D, Felton A, Holtzman DM, Mintun MA, Klunk WE, Morris JC. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009 Dec;66(12):1557-62. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Chhatwal JP, Schultz SA, McDade E, Schultz AP, Liu L, Hanseeuw BJ, Joseph-Mathurin N, Feldman R, Fitzpatrick CD, Sparks KP, Levin J, Berman SB, Renton AE, Esposito BT, Fernandez MV, Sung YJ, Lee JH, Klunk WE, Hofmann A, Noble JM, Graff-Radford N, Mori H, Salloway SM, Masters CL, Martins R, Karch CM, Xiong C, Cruchaga C, Perrin RJ, Gordon BA, Benzinger TL, Fox NC, Schofield PR, Fagan AM, Goate AM, Morris JC, Bateman RJ, Johnson KA, Sperling RA, Dominantly Inherited Alzheimer's Network Investigators. Variant-dependent heterogeneity in amyloid β burden in autosomal dominant Alzheimer's disease: cross-sectional and longitudinal analyses of an observational study. Lancet Neurol. 2022 Feb;21(2):140-152. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
In this very nice paper, Chhatwal and colleagues in the Dominantly Inherited Alzheimer’s Network report that amyloid imaging done with PiB-PET showed striking variability in participants with different PSEN1, PSEN2, and APP mutations if the mutations are assessed using different mutation grouping approaches. Despite this variability, clinical disease progression and CSF Aβ42 levels did not vary by grouping. I think these findings are interesting and are probably explained by several factors.
Once Aβ aggregates in the brain, CSF levels of Aβ42 and the Aβ42/Aβ40 ratio drop over the course of a few years. In late-onset AD and in dominantly inherited AD, this begins to occur about 20 years prior to symptom onset. A lot of data in vitro, in animal models, and in humans shows that. However, the drop in CSF Aβ42 appears to be a biochemical phenomenon not directly related to how much Aβ is aggregated in the brain per se; it more appears to relate to whether Aβ aggregation has started (more of a state change).
Further, amyloid imaging with agents such as PiB detects fibrils of Aβ. It is known in both sporadic AD, but particularly with autosomal-dominant mutations in PSEN1, PSEN2, or APP, that those mutations result in the production of differing ratios of many species of Aβ including Aβ43, 42, 40, 38, 37, and others. These differing ratios, as well as possibly other factors, determine not only whether and when Aβ aggregates, but also whether it will deposit in fibrillar or different non-fibrillar species.
It seems likely to me that while the amount of PiB-PET signal varies among the different classes of mutations at the same stage of disease, the actual amount of Aβ that is aggregated in the brain as measured biochemically may be much more similar. In the discussion, the authors point out an extreme example, whereby APPGlu693Gly (Arctic mutation) carriers do not appear to develop PiB-PET positivity. This is also seen in patients who have an amino acid deletion in the Aβ sequence. In sporadic AD, there have been cases described that are PiB-PET-negative, CSF Aβ42-positive, who at autopsy have almost exclusively non-fibrillar Aβ plaques (e.g., Cairns et al., 2009).
I think these results clearly bring out, along with prior studies, that detection of amyloid fibrils in the brain by amyloid imaging does not always reflect the location or amount of all the Aβ that is aggregated in the brain, especially when comparing different ADAD mutations. CSF Aβ42 and the Aβ42/40 ratio appear to be generally more of a “state” of Aβ aggregation marker, though other studies show that once this ratio starts to decrease, it takes a few years to reach a more stable lower value reflecting “Aβ deposition.”
Certainly, when using these different biomarkers in clinical trials, it is key to understand these differences, and depending on the therapy being tried, one needs to take into account which measure may or may not be most useful and informative.
References:
Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, Reinwald LT, Carter D, Felton A, Holtzman DM, Mintun MA, Klunk WE, Morris JC. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009 Dec;66(12):1557-62. PubMed.
BioArctic AB
Uppsala University
Uppsala University
Bioarctic
This highly interesting paper reports that, in autosomal-dominant AD, the 11C-Pittsburgh compound B (PiB)-PET signal displayed mutation-dependent variability, despite similar progression on the clinical dementia rating.
Although pathogenic variants causing autosomal-dominant AD are almost 100 percent penetrant, there is heterogeneity in levels of Aβ load between individuals. The authors investigated whether this heterogeneity was related to disease progression. The association with mutation location within PSEN1, PSEN2, or APP was also studied.
Cortical and striatal PiB-PET signals, as well as CSF Aβ42 levels, showed striking variability using either of the two mutation grouping approaches used in this study, despite similar progression on the clinical dementia rating. Highly differential temporal and regional patterns of PiB-PET signals were demonstrated, despite similar progression rates. These findings suggest that increased PiB-PET signal is generally seen in autosomal-dominant AD, but higher levels of PiB-PET signal at an individual level will not reflect more advanced disease.
A very interesting finding was that carriers of PSEN1 pathogenic variants located after codon 200 were more likely to have cerebral amyloid angiopathy (CAA), whereas those with variants before codon 200 had shorter disease duration. The disease progression was very similar in carriers of PSEN1 variants both before and after codon 200, despite the overall higher levels of PiB-PET signal in carriers of PSEN1 variants before codon 200.
Although PiB is the gold standard for imaging of brain Aβ in AD, it does not bind all Aβ species. For instance, a study by Matveev et al. (2014) showed the PiB binding site to comprise a distinct, highly insoluble subfraction of SDS-precipitated Aβ in AD frontal cortex of the brain. The majority of the extracted Aβ did not show any PiB binding when analyzed in vitro. Thus, the purified PiB binding site comprises a distinct, highly insoluble subfraction of Aβ in AD brain. These observations support the notion that high-density PiB binding is mainly seen in a discrete population of Aβ fibril-like assemblies.
Moreover, PiB binding is not detected in the inner laminar layers of the cerebral cortex (Svedberg et al. 2009), while immunoreactive Aβ clearly is present in both inner and outer layers (Braak and Braak, 1991). Thus, it is of great importance to find out why PiB fails to recognize Aβ aggregates in the inner cortical layers, and to develop future PET ligands that are more sensitive to detecting Aβ in the deeper cortical layers.
The PiB-PET signal in AD may thus not fully reflect the neurodegenerative changes associated with the clinical symptoms of cognitive decline, especially if these changes are more linked to the build-up of diffuse plaques in the inner cortical layers and to expression of soluble low-density Aβ oligomers and protofibrils.
Thus, the mutation-dependent variability seen in the report by Chhatwal et al. could reflect different mutations’ propensity to form soluble Aβ rather than dense-core plaques. An example of a PET-negative alternative is seen in the Arctic mutation carriers, E693G substitution in the APP gene, a mutation that is fully penetrant for early onset familial AD. As described by Schöll et al. (2012), Arctic mutation carriers lacked detectable PiB-PET retention, while cerebral glucose metabolism and CSF levels of Aβ42, total and phosphorylated tau were clearly pathologic. This contrasted with PSEN1 mutation carriers with high PiB retention in the cortex and the striatum in combination with abnormal glucose metabolism and positive CSF biomarkers.
Patients with sporadic AD in general show high cortical PiB retention and pathologic CSF biomarkers, as well as decreased glucose metabolism, when compared with healthy controls. The absence of PiB binding seen in Arctic mutation carriers suggests that the presence of other forms of Aβ, such as oligomers and protofibrils, is important for the pathologic processes leading to clinical AD. The lack of PiB binding was probably caused by the Aβ plaque morphology shown in Arctic mutation carriers, with diffuse, ring-like plaques (Basun et al., 2008).
Jasmeer Chhatwal and colleagues' results suggest that CSF and PET values of Aβ levels are not interchangeable and might reflect different Aβ-driven pathobiological processes. In the future in clinical trials, it may be necessary to combine PET imaging with biochemical monitoring of soluble Aβ in plasma and CSF.
PiB-PET and Aβ42 or Aβ42/40 are reasonably good surrogate markers for pathogenic aspects of AD. However, they are not pinpointing the real toxic species of Aβ, which most likely are the soluble aggregated species of Aβ, i.e., oligomers and protofibrils. This might explain the bad correlation to disease stage when using the present methods for measuring Aβ burden.
References:
Basun H, Bogdanovic N, Ingelsson M, Almkvist O, Näslund J, Axelman K, Bird TD, Nochlin D, Schellenberg GD, Wahlund LO, Lannfelt L. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008 Apr;65(4):499-505. PubMed.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
Matveev SV, Spielmann HP, Metts BM, Chen J, Onono F, Zhu H, Scheff SW, Walker LC, LeVine H 3rd. A distinct subfraction of Aβ is responsible for the high-affinity Pittsburgh compound B-binding site in Alzheimer's disease brain. J Neurochem. 2014 Nov;131(3):356-68. Epub 2014 Jul 28 PubMed.
Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, Almkvist O, Graff C, Nordberg A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012 Jul 17;79(3):229-36. PubMed.
Svedberg MM, Hall H, Hellström-Lindahl E, Estrada S, Guan Z, Nordberg A, Långström B. [(11)C]PIB-amyloid binding and levels of Abeta40 and Abeta42 in postmortem brain tissue from Alzheimer patients. Neurochem Int. 2009 May-Jun;54(5-6):347-57. PubMed.
Lund University
I want to congratulate the authors on a very intriguing and comprehensive study showing that the regional PiB PET retention differs quite substantially between different AD-associated APP and PSEN mutations in relation to the expected years to symptom onset. The heterogeneity in Aβ-PET retention has very important implications for anti-amyloid trials in ADAD when using Aβ-PET to determine target engagement and when correlating reductions in Aβ-PET signal with clinical outcome measures.
Interestingly, the reduction in CSF Aβ42 levels seems to be more consistent between different APP and PSEN mutations. It is well known that individuals with the Arctic APP mutation exhibit diffuse (“Congo red negative”) Aβ-plaques, in addition to tau-containing neuropil threads and tangles as well as neurodegeneration (Kalimo et al., 2013). In these cases, PiB retention is very low, but CSF Aβ42 and P-tau levels are both clearly abnormal (Schöll et al., 2012). Consequently, it would be interesting to determine whether the results in the present study are explained by regional differences in the ratios between diffuse and cored Aβ plaques between different APP and PSEN mutations, where PiB preferentially binds to cored plaques, but reduced CSF Aβ42 levels are likely to be associated with both types of plaques.
Finally, the current results imply to me that CSF Aβ42 should complement Aβ-PET as an outcome in ADAD trials when evaluating anti-amyloid therapies.
References:
Kalimo H, Lalowski M, Bogdanovic N, Philipson O, Bird TD, Nochlin D, Schellenberg GD, Brundin R, Olofsson T, Soliymani R, Baumann M, Wirths O, Bayer TA, Nilsson LN, Basun H, Lannfelt L, Ingelsson M. The Arctic AβPP mutation leads to Alzheimer's disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol Commun. 2013 Sep 10;1:60. PubMed.
Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, Almkvist O, Graff C, Nordberg A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012 Jul 17;79(3):229-36. PubMed.
Make a Comment
To make a comment you must login or register.