In Progressive Supranuclear Palsy, Filamin-A Goads Tau Aggregation
Quick Links
In Alzheimer's disease, aggregation of Aβ precedes tau aggregation, but in primary tauopathies, the upstream instigators are shrouded in mystery. A study published May 25 in Science Advances casts light on a potential troublemaker in progressive supranuclear palsy (PSP). Researchers led by Masahisa Katsuno, of Nagoya University Graduate School of Medicine in Japan, spotted filamin-A mingling with tau aggregates in the brains of people who had died with the disease. What’s more, they identified rare variants in the filamin gene among PSP cases, even a duplication of the gene in a set of twins who had died from the disease. In cultured cells, filamin-A coordinated with actin to promote tau aggregation, and in transgenic mice overexpressing the human version of the actin-binding protein, tau aggregates formed within neurons. The findings highlight a potential mechanism afoot in PSP, and may point to therapeutic targets.
- Filamin-A was found mingling with tau tangles in the brains of people with PSP.
- Rare variants in FLNA detected among PSP cases, including a set of twins.
- Overexpression of FLNA drives tau aggregation in transgenic mice.
Considered a primary tauopathy, PSP is marked by inclusions consisting of four-repeat tau, which contains all four microtubule-binding domains. In contrast, the tangles that form in AD comprise both 3R- and 4R- isoforms. In PSP, tau aggregates form in neurons, oligodendrocytes, and astrocytes. In fact, so-called “tufted astrocytes” are a hallmark of the disease. However, the majority of people with PSP harbor no known pathogenic mutations. Even among familial cases, only some carry a pathogenic mutation in the tau gene. As such, the mechanisms that kick off tau aggregation in most PSP cases remain unclear.
To hunt for possible instigators of tau aggregation in PSP, first author Koyo Tsujikawa and colleagues isolated insoluble protein fractions from the brains of 11 people with PSP, 10 with corticobasal degeneration (CBD), 10 with AD, six with Parkinson’s disease (PD), five with dementia with Lewy bodies (DLB), and five normal controls. Using mass spectrometry, they identified large aggregates of tau containing filamin-A (FLNA) and several other proteins in the PSP samples. Via immunofluorescence, the researchers also spotted FLNA cozying up with tau aggregates in neurons and tufted astrocytes in brain sections from people with PSP, but not in tissue from people who had died with other tauopathies, including corticobasal degeneration, Pick’s disease, or Alzheimer’s disease (see image below). The findings implied that in PSP, FLNA appeared to have a potentially toxic relationship with tau.
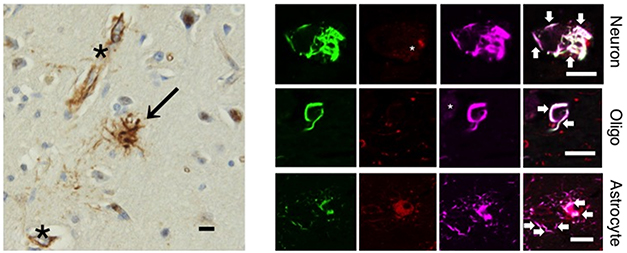
Filamin Filaments? Immunohistochemistry (left) from the frontal cortex of a person with PSP reveals filamin-A deposits in tufted astrocytes (arrow) and blood vessels (asterisk). By immunofluorescence (right), filamin-A (purple) co-localizes with hyperphosphorylated tau (green) in neurons, oligodendrocytes, and astrocytes. [Courtesy of Tsujikawa et al., Science Advances, 2022.]
To hunt down other FLNA variants that associate with PSP, the scientists used genetic samples from the Japanese Alzheimer’s Disease Neuroimaging Initiative (J-ADNI), and Japanese Longitudinal Biomarker Study in PSP and CBD (JALPAC) Consortium, amassing 312 people with PSP, 71 with CBD, 141 with AD, and 499 controls. Twelve PSP cases carried rare variants in FLNA, six were missense mutations, and one a splicing variant. However, this was no slam dunk. Other rare variants in FLNA cropped up in two cases of CBD, in three cases of AD, and even in five controls. Still, PSP had the highest rate of rare variants in FLNA.
The researchers got another hint that the actin-binding protein might play a role in the disease when they sequenced the exomes of a Japanese family affected by PSP, but sans pathogenic tau mutations. Searching for the causal mutation in monozygotic twins in this family, who had both died of PSP at age 67, the researchers discovered that both twins carried an extra copy of Xq28—a segment of the X chromosome that includes FLNA and 15 other genes. None of 513 healthy controls, including all but one other person in the family, had this duplication variant. A sister of the twins did carry the FLNA duplication, but did not develop the disease, possibly due to a dramatically skewed pattern of X inactivation that snuffed out expression of the problem variant.
The researchers measured elevated levels of FLNA protein in the brains of the twins relative to controls. To study how this might influence tau, the researchers grew lymphoblast cell lines. In cells from the twins, both FLNA and tau levels rose relative to levels in control cells. Knocking down FLNA lowered tau, suggesting that the actin-binding protein somehow stokes tau levels.
How might FLNA influence tau expression or aggregation? The researchers employed a barrage of biochemical experiments in cultured cells to find out. In a nutshell, they report that FLNA directly interacts with 4R-tau, stabilizing the microtubule-binding protein and enhancing its hyperphosphorylation. Actin polymerization appeared to play a role in this, because the researchers found that several of the missense variants identified in PSP enhanced FlLNA’s ability to promote actin polymerization.
Could this boosted actin polymerization promote 4R-tau aggregation? While most missense variants that boosted actin polymerization also elevated 4R-tau levels in cultured cells, the p.Ala39Gly FLNA mutant, which cannot polymerize actin, did not. This was also true in the brains of mice transduced with FLNA. Overexpression of wild-type FLNA boosted actin polymerization and tau aggregation, while overexpression of pAla39Gly mutant did not. Tau has several actin-binding domains, and previous studies have implicated interactions between tau and actin in neurodegeneration (Jan 2007 news; Bardai et al., 2018).
Lastly, the researchers investigated how excess FLNA in the mouse brain would influence endogenous tau. Whether induced by an adenoviral vector expressing human FLNA, or the full-length human gene inserted into the mouse genome, FLNA overexpression revved the accumulation of 4R-tau in the mouse brain. In the transgenic mice, deposits of 4R-tau popped up in regions where human FLNA was overexpressed the most—in neurons of the hippocampus and deeper cortical layers. Hyperphosphorylated tau was spotted within some of these deposits. Not typical of PSP, relatively few glial cells harbored 4R-tau inclusions and there were no tufted astrocytes; this may be because transgenic expression of FLNA was much lower in glial cells than in neurons.
To the authors, the findings suggest that FLNA exacerbates a pathological interaction between tau and F-actin. It is unclear why this would be specific to PSP, though the authors noted that 4R-tau contains more actin-binding domains than 3R-tau. It also remains unclear if filamin-A levels are up in the brains of people with sporadic PSP, who have no underlying mutation in the gene.
If filamin-A sounds familiar, it could be because of simufilam, a small-molecule drug in clinical development for AD. Simufilam reportedly binds to FLNA, and blocks interactions between Aβ, the α7 nicotinic acetylcholine receptor and TLR-4 (Jul 2012 news; Wang et al., 2017). The drug’s developer, Cassava Sciences, is the subject of ongoing investigations into claims of data manipulation.
Tsujikawa and colleagues do not argue for developing simufilam for PSP, which is not marked by Aβ pathology. They did highlight a recent proteomics study in a mouse model of AD indicating that memantine—an approved drug for AD—nearly halved expression of FLNA in the hippocampus, suggesting it could possibly dampen FLNA-induced tau aggregation (Zhou et al., 2019). —Jessica Shugart
References
News Citations
- New Takes on Tau: Does It Stabilize Actin, Flag Neurogenesis?
- Novel Drug Knocks Aβ Off Synapses, Reduces Toxicity
Therapeutics Citations
Paper Citations
- Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, Feany MB. A Conserved Cytoskeletal Signaling Cascade Mediates Neurotoxicity of FTDP-17 Tau Mutations In Vivo. J Neurosci. 2018 Jan 3;38(1):108-119. Epub 2017 Nov 14 PubMed.
- Wang HY, Lee KC, Pei Z, Khan A, Bakshi K, Burns LH. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer's disease pathogenesis. Neurobiol Aging. 2017 Jul;55:99-114. Epub 2017 Mar 31 PubMed.
- Zhou X, Wang L, Xiao W, Su Z, Zheng C, Zhang Z, Wang Y, Xu B, Yang X, Hoi MP. Memantine Improves Cognitive Function and Alters Hippocampal and Cortical Proteome in Triple Transgenic Mouse Model of Alzheimer's Disease. Exp Neurobiol. 2019 Jun;28(3):390-403. Epub 2019 Jun 26 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Tsujikawa K, Hamanaka K, Riku Y, Hattori Y, Hara N, Iguchi Y, Ishigaki S, Hashizume A, Miyatake S, Mitsuhashi S, Miyazaki Y, Kataoka M, Jiayi L, Yasui K, Kuru S, Koike H, Kobayashi K, Sahara N, Ozaki N, Yoshida M, Kakita A, Saito Y, Iwasaki Y, Miyashita A, Iwatsubo T, Japanese Alzheimer’s Disease Neuroimaging Initiative (J-ADNI), Ikeuchi T, Japanese Longitudinal Biomarker Study in PSP and CBD (JALPAC) Consortium, Miyata T, Sobue G, Matsumoto N, Sahashi K, Katsuno M. Actin-binding protein filamin-A drives tau aggregation and contributes to progressive supranuclear palsy pathology. Sci Adv. 2022 May 27;8(21):eabm5029. Epub 2022 May 25 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.