Alzheimer’s in a Dish? Aβ Stokes Tau Pathology in Third Dimension
Quick Links
Don your IMAX glasses. Researchers have taken a step closer to modeling Alzheimer’s disease in a dish by using a three-dimensional gel matrix. As described in Nature on October 12, researchers led by Doo Yeon Kim and Rudolph Tanzi at Harvard Medical School in Charlestown, Massachusetts, constructed a human neural cell culture system that develops both amyloid plaques and neurofibrillary tangles. Neurons in the three-dimensional culture overexpress disease-associated mutations that ramp up Aβ production, and tau pathology ensues. The system is the first to reveal a direct causal link between Aβ production and tau pathologies, Tanzi told Alzforum, and may serve as a handy tool to screen drugs that target them.
“It’s really beautiful work, and is the first demonstration of a human model of AD,” commented Terrence Town of the University of Southern California in Los Angeles. “The tau pathology appears to be consequent to the amyloid pathology, and that’s key because it supports the amyloid hypothesis.”
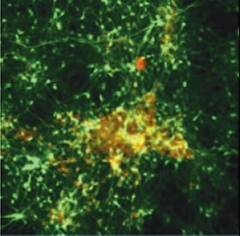
Deposition in a Dish.
After six weeks in three-dimensional gel culture, neurons (green) expressing familial AD mutations form extracellular amyloid deposits (red/yellow). [Image courtesy of Choi et al., Nature 2014]
The amyloid hypothesis was born 30 years ago, when researchers zeroed in on the identity of the amyloid fibrils that dominated AD brains, and eventually linked familial forms of the disease to APP (see Glenner and Wong, 1984; Yankner et al., 1989; and Levy et al., 1990). The core tenet of the hypothesis is that Aβ drives AD pathology, including the development of neurofibrillary tau tangles (see Hardy and Selkoe, 2002), but modeling this in any one system has been an elusive goal. Mice engineered to ramp up Aβ production and accumulate amyloid develop no tau pathology unless other transgenes are added. Mice expressing mutant forms of tau do develop tauopathy (for example, the Tau P301S and 3xTg lines), but the consensus is that these model frontotemporal dementia rather than AD (see Chin, 2011).
One hint of Aβ being upstream of tau in mice, too, came from a study that detected an age-related tau increase in the CSF of aging mice that express APP mutations (Maia et al., 2013). Town’s lab developed an APP-based rat model that develops tauopathy, likely because rats, unlike mice, express six isoforms of tau—as do humans—but it is not yet widely used (see Cohen et al., 2013).
Cell culture models have fared no better. A range of Aβ oligomers can be produced, but larger insoluble amyloid plaques do not form. “To date, we have not had a model for AD that makes plaques in a dish, let alone a model where plaques actually lead to tangles,” Tanzi told Alzforum.
In an attempt to unite Aβ and tau pathologies into a single model, co-first authors Se Hoon Choi and Young Hye Kim started with a human two-dimensional neuron culture and then added some structure. They selected a commercially available human neural progenitor cell line—monoclonal cells derived from fetal neural stem cells—as the star of their model. They infected the progenitor cells with lentiviral vectors carrying human APP harboring the Swedish and London familial AD (FAD) mutations, with or without the FAD-linked PSEN1ΔE9. After six weeks in culture conditions that promote neuronal differentiation, the neurons overexpressing mutant APP pumped out 19-fold more Aβ42 than control cells, and co-expression of PSEN1Δ9 boosted Aβ production another fivefold.
The researchers hypothesized that if Aβ did not wander away in the culture medium, it might form deposits. To corral Aβ, the researchers grew the neural cultures within a three-dimensional support gel chock-full of brain extracellular matrix proteins. In this structured environment, the progenitor cells differentiated into more mature neurons than those in the two-dimensional culture system, and also expressed higher levels of 4-repeat adult tau isoforms, which are thought to be essential for the development of tauopathy. Six weeks later, confocal microscopy revealed large amyloid deposits lurking in three-dimensional cultures that expressed FAD mutants. The aggregates reacted with several different Aβ-specific antibodies, as well as Congo Red and AmyoGlo, an amyloid-specific dye. Treatment with β- or γ-secretase inhibitors dramatically reduced these deposits.
Tau pathology also developed. By six weeks in culture, western blots revealed an accumulation of soluble and insoluble hyperphosphorylated tau. By immunohistochemistry, the researchers noted some neurons harbored highly elevated levels of phosphorylated tau. They also displayed odd morphologies, such as neurites with the same beaded processes that have been found in the brains of AD patients. To the researchers’ surprise, treatment of the cultures with either β- or γ-secretase inhibitors decreased levels of phospho-tau in extracts as well as the number of cells accumulating these isoforms in culture.
In cultures enriched for cells that expressed the most APP and PSEN1, the researchers found an abundance of phosphorylated tau in neurites and neuronal cell bodies. High molecular weight, hyperphosphorylated forms of tau dominated insoluble fractions (see image below). When the researchers looked at these aggregates with transmission electron microscopy, they observed filaments strikingly similar to those found in AD. Treatment with β- or γ-secretase inhibitors dramatically reduced production of these tau isoforms in the neurons, suggesting a link between Aβ production and tauopathy. By 10 weeks in culture, the neurons contained inclusions detectable by silver staining, a classic measure of tau tangles.
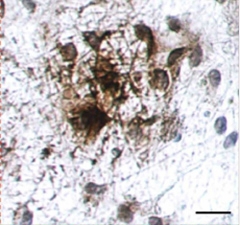
Tau Takeover.
Gobs of hyperphosphorylated tau (brown) accumulate in cell bodies and neurites of neurons expressing familial AD mutations, after six weeks in three-dimensional gel culture. [Image courtesy of Choi et al., Nature 2014.]
Interestingly, when the researchers treated cultures with an inhibitor of GSK3β, a tau kinase implicated in AD, phosphorylated tau, but not Aβ, fell dramatically. This indicated that the kinase does indeed mediate Aβ-triggered tau pathology and that tau lies downstream of Aβ.
While these cultures may have captured the pathological cascade that can occur among neurons in AD brains, they did not account for the potential role of other cell types, such as microglia and astrocytes. That Aβ induced tau pathology in the absence of immune cells, such as microglia, suggests that inflammation may not be required for Aβ to drive tauopathy, Tanzi said. Such an intermediary step had been proposed by others as part of the amyloid cascade hypothesis. Kim added that the role of microglia should not be overlooked, however. “We cannot discount the impact of microglia on this process, because we may see increases in pathology when we add those cells in,” he said. Kim and Tanzi said they plan to more carefully scrutinize the potential role of neuroinflammation by including microglia in their cultures, among other modifications. “We are just at the beginning,” Tanzi said.
Town agreed that the model was limited in its current form, but could be expanded to include neuroinflammatory or vascular components in the future. “It’s very hard to get one model to study everything,” he said.
The culture model will also allow the researchers to more carefully tease out the links between Aβ production and tauopathy, including how GSK3β serves as an intermediary, which species of Aβ drive tauopathy, and how these factors may influence neurodegeneration, the authors said. Researchers are still not sure which form of Aβ is most toxic. “Given that this is up for debate right now, it is very valuable to have a system where you can actually manipulate plaques and tangles,” commented Asa Abeliovich of Columbia University in New York, who was not involved in the work. Abeliovich was impressed with the level of tau pathology the researchers observed in their system. “There’s a tremendous need to have accurate disease models of neurodegeneration, and it’s tremendously difficult to come up with,” he said. “I like the simplicity of their system.”
Would such a culture model work with AD patient-derived cells, such as iPSCs? Generating amyloid and tau pathology in such models would be difficult without overexpression, Tanzi said. “If you want to recapitulate in two to three months pathology that takes decades to form in the human brain, you most likely have to rely on overexpression,” he said. Other researchers agreed.
Tanzi sees the culture model as a boon to drug discovery, particularly for drugs aimed at knocking down amyloid and/or tau pathologies. “This cell culture model could make drug screening 10 times faster and 10 times cheaper than testing in animal models,” he speculated.
David Holtzman of Washington University in St. Louis, who was not involved in the work, agreed that the culture system would make for a good screen. “This is now a model system to potentially rapidly test new treatments targeting Aβ, tau, or even other targets as new treatments,” he wrote to Alzforum (see full comment below).—Jessica Shugart
References
Research Models Citations
Mutations Citations
Paper Citations
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984 May 16;120(3):885-90. PubMed.
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989 Jul 28;245(4916):417-20. PubMed.
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990 Jun 1;248(4959):1124-6. PubMed.
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353-6. PubMed.
- Chin J. Selecting a mouse model of Alzheimer's disease. Methods Mol Biol. 2011;670:169-89. PubMed.
- Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M. Changes in amyloid-β and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013 Jul 17;5(194):194re2. PubMed.
- Cohen RM, Rezai-Zadeh K, Weitz TM, Rentsendorj A, Gate D, Spivak I, Bholat Y, Vasilevko V, Glabe CG, Breunig JJ, Rakic P, Davtyan H, Agadjanyan MG, Kepe V, Barrio JR, Bannykh S, Szekely CA, Pechnick RN, Town T. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013 Apr 10;33(15):6245-56. PubMed.
Further Reading
Papers
- Doege CA, Abeliovich A. Dementia in a dish. Biol Psychiatry. 2014 Apr 1;75(7):558-64. Epub 2014 Jan 27 PubMed.
- Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem Pharmacol. 2014 Apr 15;88(4):508-16. Epub 2014 Jan 21 PubMed.
Primary Papers
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014 Nov 13;515(7526):274-8. Epub 2014 Oct 12 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
It is clear from genetics, biochemistry, and both animal studies and human studies that Aβ and tau are both important in causing Alzheimer’s disease.
There is also a lot of suggestive data that the accumulation of Aβ in the brain somehow drives the accumulation and spread of tau aggregation and pathology in the brain, which is important in neurodegeneration. What is lacking in the field is a model in cell culture that recapitulates these key features of disease. In this paper by Choi et al., using human neural progenitor cells, the researchers show that by growing them in a three-dimensional matrix, there is not just increased levels of Aβ42 being produced, but there is clear-cut amyloid-plaque formation. This is the first time that I am aware of this being shown in cultured cells. Equally important, a form of tau that is more prone to aggregate, called 4-repeat tau, is produced by these human neural cells and in the presence of amyloid plaques, forms hyperphosphorylated, detergent-insoluble aggregates. This detergent-insoluble form of tau is what accumulates in Alzheimer’s disease and related disorders and is a signature of these diseases. This new model and findings are important for the following reasons:
1. It is the first time that Aβ plaques and neurofibrillary tangles containing tau form spontaneously in cultured cells;
2. The data strongly suggest that Aβ aggregation can directly lead to tau aggregation when using human cells;
3. This new system provides a nice model to determine how Aβ drives tau pathology;
4. This is now a model system to potentially rapidly test new treatments targeting Aβ, tau, or even other targets as new treatments.
University of Texas, Southwestern Medical Center
The significance of this paper could be enormous if it facilitates discovery of mechanisms that link Aβ production/accumulation to tau pathology. In this model, the authors have shown Aβ accumulation, and tau becomes insoluble and hyperphosphorylated. This confirms and extends Shi et al., 2012, which reported similar findings in a different in vitro system.
To further explore whether this system recapitulates AD in a dish, the identity of the neurofibrillary tangles could be probed at greater depth methodologically. The EM image provided is a start, but immuno-EM requires careful use of negative controls and blinded analyses to be sure. My experience in searching for fibrils under EM has been that it is not too hard to find fibril-like structures in the cell. It would be helpful to rule out that background staining by antibodies has labeled some other fibril-like cell structure. Therefore a next step in replicating this potentially very important finding would be to use unbiased metrics to analyze EM images. Another, simpler, step to ascertain that this system indeed accumulates intracellular amyloids is to perform amyloid stains on the cell culture.
This study is a great start, and now requires replication in other labs.
References:
Shi Y, Kirwan P, Smith J, Maclean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med. 2012 Mar 7;4(124):124ra29. PubMed.
Institute of Neurology, UCL
This paper is an undoubted step forward in the extent of both amyloid and tau pathology. It is not correct, however, to say that this is entirely novel. Shi et al., 2012, showed both Aβ deposition and tau pathology in iPSC-derived neuron cultures derived from Down syndrome (i.e. in the context of physiological levels of expression).
References:
Shi Y, Kirwan P, Smith J, Maclean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med. 2012 Mar 7;4(124):124ra29. PubMed.
KULeuven
This paper reports a long-overdue in vitro approach to a long-standing problem.
Nevertheless, it is important to keep in mind that neurons in culture are in a permanently stressed situation that does not reflect normal neurons in the brain but could be construed to reflect stress as in AD. Moreover, the lentiviral transfection used to drive transgene expression to high levels stresses the cultured neurons even further. As important, an indisputable limitation of most transgenic models applies here, too, in that the cultured neurons express the APP Swedish and London double mutant plus the Δ9 presenilin mutant, and massively produce mutant APP and amyloid peptides.
Even so, the outcome of combined amyloid and tau pathology appears most interesting for the amyloid cascade hypothesis. It replicates in vitro the important argument that even the most early onset, obligatory amyloid-driven familial forms of AD suffer tauopathy besides amyloid. The presence of both, of course, was Alois Alzheimer’s original definition of the disease based on Auguste Deter’s familial case, and it still holds. That said, this study still only speaks to FAD and does not model the causes of late-onset sporadic AD.
The tauopathy outcome in this study conforms to our findings in APP-V717I (London) mouse brain that amyloid activates GSK3 and increases the phosphorylation of endogenous mouse protein tau from the age of three to six months, prior to amyloid deposition (Terwel et al., 2008). One cannot fully assess the extent and authenticity of tauopathy in this culture system from the data presented. We all know that is technically complex, even more in neuronal cultures than in brain. It will require western blotting and immunohistochemistry for other robust phospho-epitopes in addition to AT8 and PHF1, besides more extensive EM-ultrastructural analysis.
An important point raised by this paper is the renewed implication of GSK3—either α or β or both—in AD. The study reinforces the argument, generally accepted in AD research, that GSK3 is the primary, though by no means the only, kinase that phosphorylates tau in vivo. GSK3 profoundly aggravates tauopathy and affects many neuronal functions and networks (Spittaels et al., 2000; Terwel et al., 2005, 2008; Crespo-Biel et al., 2014; Maurin et al., 2014).
However, despite very intensive efforts from small and big pharma, GSK3 inhibitors have not yet made it into the clinic, and in my opinion they never will. The major reason is that GSK3 is expressed in most body tissues and involved in a great variety of signaling pathways, most of which are basic and essential for physiological functions. In brain, GSK3 is important for pre- and post-synaptic mechanisms underlying learning and memory, involving LTP and LTD, that we and many other groups are still trying to dissect (e.g., Ochs et al., 2014). Much as I welcome this renewed enthusiasm in our continued fight against this dreadful disease, GSK3 does not appear as a feasible drug target.
As always, I am eagerly looking forward to follow-up studies to this new and interesting model.
References:
Terwel D, Muyllaert D, Dewachter I, Borghgraef P, Croes S, Devijver H, Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008 Mar;172(3):786-98. PubMed.
Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000 Dec 29;275(52):41340-9. PubMed.
Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005 Feb 4;280(5):3963-73. PubMed.
Crespo-Biel N, Theunis C, Borghgraef P, Lechat B, Devijver H, Maurin H, Van Leuven F. Phosphorylation of protein Tau by GSK3β prolongs survival of bigenic Tau.P301L×GSK3β mice by delaying brainstem tauopathy. Neurobiol Dis. 2014 Jul;67:119-32. Epub 2014 Apr 2 PubMed.
Maurin H, Chong SA, Kraev I, Davies H, Kremer A, Seymour CM, Lechat B, Jaworski T, Borghgraef P, Devijver H, Callewaert G, Stewart MG, Van Leuven F. Early structural and functional defects in synapses and myelinated axons in stratum lacunosum moleculare in two preclinical models for tauopathy. PLoS One. 2014;9(2):e87605. Epub 2014 Feb 3 PubMed.
Ochs SM, Dorostkar MM, Aramuni G, Schön C, Filser S, Pöschl J, Kremer A, Van Leuven F, Ovsepian SV, Herms J. Loss of neuronal GSK3β reduces dendritic spine stability and attenuates excitatory synaptic transmission via β-catenin. Mol Psychiatry. 2014 Jun 10; PubMed.
Case Western Reserve University
The impediment to making progress in AD research and finding a cure is not the lack of success of the traditional reductionist approach, where a complex phenomenon is broken down into individual components and studied to better understand the phenomenon. We fail in the “synthetic” approach, where individual components are put together to gain a more complete understanding of disease pathogenesis at the patient level.
This study—the disease in a dish—is a brilliant piece of in vitro experimentation. The excitement it has generated will be fully manifested if it resonates with clinical findings. Unfortunately, here we are on shakier ground:
1. In hundreds of brain autopsies, Braak and colleagues observed spatial and temporal dissociation between amyloid pathology and tau pathology, with the latter appearing decades prior to and in a different location in the brain than the former. The Lille group also demonstrated that tau pathology progresses from the limbic system to the neocortex, while amyloid pathology moves in the opposite direction. Thus, the in vitro insistence that amyloid pathology causes tau pathology is discordant with the fundamental in vivo clinical observations in late-onset AD.
2. Brain imaging studies have shown that areas of amyloid deposits do not necessarily coincide with those of hypometabolism, which in turn do not coincide with those of brain atrophy. The simplest interpretation of the multimodal imaging data is that amyloid deposits are not toxic to nearby neurons in vivo (indeed, neurons in the Choi et al. study still looked healthy after six to eight weeks around amyloid deposits). If so, the potential of this new system for understanding the “pathogenic mechanisms of AD” might be overestimated.
3. We cannot ignore that the SNAP phenomenon (individuals neurologically diagnosed as preclinical AD but lacking amyloid deposits by brain imaging) is observed in 20-25 percent of the population. While it is true that nosologically these patients may not be classified as AD as defined by Alois Alzheimer, it is clear that we can have symptoms of early AD in the absence of amyloid deposits.
4. The simplicity of the three-dimensional cell culture system, which will allow screening of thousands of drug compounds quickly and inexpensively, may prove to be limiting because of the lack of a blood-brain barrier. Many drugs that showed promise in vitro and/or in mouse models are thought to have failed in clinical trials because of the impenetrable blood-brain barrier.
This is not to rain on the amyloid parade. I am awed by the experimental manipulations described here. However, it is important to put these findings in context and provide a balanced view. As a field, we are better off if we underpromise and overdeliver.
References:
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011 Nov;70(11):960-9. PubMed.
University of Edinburgh
This study is a lovely confirmation of the cascade from amyloid to tau pathology in a human cellular system, and the first cell culture system that I have heard of to develop both plaques and tangles. While this new system is not a perfect recapitulation of sporadic Alzheimer's (as it relies on overexpression of proteins), having human cells develop the pathological hallmarks of Alzheimer's disease in a dish in a relatively short time frame is a leap forward for the field. This will allow rapid tests of new treatments in cells that have the same machinery as human patients, hopefully making these tests an accurate predictor of whether the treatments will work in people.
Make a Comment
To make a comment you must login or register.