Past Webinar
Calcium Signaling Deficit in the Origin of Alzheimer's Disease
Quick Links
Introduction
Live discussion and panel of participants scheduled for 7 September 2001.
The live chat and panel of participants scheduled for 7 September 2001 has been postponed until further notice.
Background
Background Text
By Ming Chen
Much of the research on Alzheimer's disease currently is devoted to the issue of what plaques and tangles do in the brain. However, I would argue that it may be more important to ask,"where do they come from?" This should be the central issue in AD research.
Conceptually, there are two groups of substances in our body which are associated with diseases. The first group includes cancer growth, HIV proliferation, etc, which are caused by pathogens. The second group includes "aging markers" such as hair graying, skin wrinkling, tooth loss, cholesterol deposits, or gallstone.
If plaques and tangles are like cancer or virus, then we would need to look for pathogens that are independent from normal metabolic processes. But if plaques and tangles are analogous to cholesterol, gallstone or cataract deposition, then the direction of our quest would be different, because these aging markers perhaps all originate from aging-associated inefficiencies of normal metabolism. For example, cholesterol accumulation is perhaps the result of inefficiency of its normal degradation, and gallstone is due to slowdown of the mineral clearance, and so on. In these cases, age-related changes in the normal metabolic pathways, rather than a "pathogen," would be key to understanding these disorders.
For a long time, we, like many others, had taken the former scenario as granted because it is so widely believed. Recently, however, based on our experimental findings and a comprehensive review of literature, we were finally led, to our surprise, to the conclusion that the latter scenario is the case. This change did not occur in one day, but step by step through a series of publications over the past several years:
1. We first found that alpha-secretase, the protease responsible for APP normal processing, is a Ca2+-dependent protease (1), possibly calpain (5, 9), a well-known Ca2+-dependent protease. This finding was made several years ago, but its publication proved difficult because it contradicts other reports. This finding has not established the identity of alpha-secretase, but suggested a clue for the regulatory mechanism of this enzyme.2. Following this clue, we reasoned that if alpha-secretase is regulated by Ca2+, then many agents that have been found to enhance soluble APP (APPs) secretion would also activate Ca2+. Conversely, agents that reduce APPs would have an opposite effect on Ca2+. We immediately checked this possibility by sorting through many reports and found that it is essentially the case (2). Indeed, this finding should not be a surprise because it is well known that protein secretion, an essential part of cell metabolism, is generally under the control of Ca2+ (3, 6).
3. Thus, the concept that APP alpha-processing is a Ca2+-dependent process is supported not only by our finding, but also by an unusually high consistency of many other stuides. Therefore, we reasoned that if alpha-secretase reduces its activity during aging, then APP will accumulate. This, in turn, will provide excessive substrate for beta- and gamma-secretases to overproduce A-beta (2, 7). The two pathways compete for the same APP pool, thus decrease in one must increase the other, in much the same way as the increased deposition of cholesterol must come from the reduced normally-degraded molecules. When a protein becomes deposited in normal aging, the first suspect should be the possible failure in its normal degradation pathway (protein turn-over slowdown).
4. Why will alpha-secretase reduce its activity? Because aging is a process between dynamic life (young) and death (full-stop of metabolisms). So, during this process, most metabolic activities should diminish (6). Ca2+ signaling, like any other essential biochemical pathways (such as cAMP signaling, energy metabolism, protein secretion, and cholestrol catabolism) must be down-regulated during aging.
5. Surprisingly, this model may also explain tangle formation. It is well-known that tau is normally degraded by calpain (indeed, many cytoskeleton proteins are also normally degraded by calpain)(3). Tau is also normally dephosphorylated by phosphatases including calcineurin (Ca2+-dependent phosphase) (2, 6, 7). So, when Ca2+ signal is reduced, tau will accumulate and at the same time become hyperphosphorylated.
6. More importantly, aging and AD are characterized not only by plaques and tangles, but also by a progressive neurotransmission decline. What factor controls neurotransmission? Textbooks tells us that this factor is Ca2+ alone. So, a Ca2+ signaling deficit can explain all three hallmarks, together with the question of why they appear in everybody and at about the same time in life.
7. However, this view collides immediately with a current theory, i.e., "calcium overload" hypothesis, which claims that intracellular Ca2+ levels are increasing throughout aging, leading to cell death. But is is known today that Ca2+ exerts its effects not through "steady-state level" changes (like water levels in swimming pool, as conventionally conceived), but rather through rapid changes in its spike frequency and amplitude (like radio waves) (8). Ca2+ spikes are highly energy-dependent (6) and energy levels must decrease during aging. So, Ca2+ spike frequency will naturally reduce during aging. Intriguingly, the reduced frequency means a "prolonged Ca2+ stay" in the cytosol. Because the spikes in vivo occurs within sub-millisecond, if Ca2+ is measured in second or minute time intervals (as most reports do), it can appear as a slightly "increased level". But such an apparent increase actually means a decreased signaling potency (8).
Although the proposed roles of Ca2+ in plaque and tangle formation is debatable, our model emphasizes one basic point: plaques, tangles and mild memory deficits in most people (except for inherited cases) occur as a result of inefficient NORMAL metabolic pathways and thus are NATURAL events during aging like many other aging markers, but not caused by any pathogens or metabolic "mistakes" like in cancer growth.
This hypothesis predicts:
1. Ca2+ activators (estrogens, nerve growth factors, etc.) together with physical exercise will have neuroprotective effects in the elderly, because they all promote physiological metabolisms. Hence, they will not only increase the production of soluble APP (APPs), but also reduce A-beta (2, 3). This view is similar to the concept of promoting normal metabolism in order to reduce cholesterol deposits, gallstones, osteoporosis, muscle atrophy and other conditions associated with aging.
2. Conversely, animal models for AD can be generated by prolonged use of Ca2+ antagonists or energy metabolism inhibitors (4, 6), or by reducing the intrinsic Ca2+ activators (such as NGF).
3. Presenilins most likely act as Ca2+ channels in vivo (their structures are highly channel-like) and their mutations will REDUCE the Ca2+ channeling ability (2, 3). This will decrease Ca2+ spike FREQUENCY and amplitude, and thus increase A-beta by inactivating alpha-secretase, and also reduce normal degradation of tau and normal neurotranmission altogether. These predictions can be experimentally verified.
References
1. Chen M. Alzheimer's alpha-secretase may be a calcium-dependent protease. FEBS Lett. 1997 Nov 10;417(2):163-7. Abstract.
2. Chen M. The Alzheimer's plaques, tangles and memory deficits may have a common origin. Part I: a calcium deficit hypothesis. Front Biosci. 1998 May 11;3:a27-31. Abstract.
3. Chen M. The Alzheimer's plaques, tangles and memory deficits may have a common origin. Part II: therapeutic rationale. Front Biosci. 1998 Jun 8;3:A32-7. Abstract.
4. Chen M. The Alzheimer's plaques, tangles and memory deficits may have a common origin. Part III: animal model. Front Biosci. 1998 Jun 17;3:A47-51. Abstract.
5. Chen M, Fernandez HL. The Alzheimer's plaques, tangles and memory deficits may have a common origin. Part IV: can calpain act as alpha-secretase? Front Biosci. 1998 Dec 15;3:A66-75. Abstract.
6. Chen M, Fernandez HL. The Alzheimer's plaques, tangles and memory deficits may have a common origin. Part V: why is Ca2+ signal lower in the disease? Front Biosci. 1999 Apr 1;4:A9-15. Abstract.
7. Chen M. Do the intracellular calcium states in Alzheimer disease need to be revisited? J Neuropathol Exp Neurol. 1999 Mar;58(3):310-1. Abstract.
8. Chen M, Fernandez HL. Ca2+ signaling down-regulation in ageing and Alzheimer's disease: why is Ca2+ so difficult to measure? Cell Calcium. 1999 Sep-Oct;26(3-4):149-54. Abstract.
9. Chen M, Durr J, Fernandez HL. Possible role of calpain in normal processing of beta-amyloid precursor protein in human platelets.Biochem Biophys Res Commun. 2000 Jun 24;273(1):170-5. Abstract.
The Following Three Figures Summarize our "Ca2+ deficit" Hypotheses
Fig. 1. Two different "Ca2+ deficit" hypotheses.
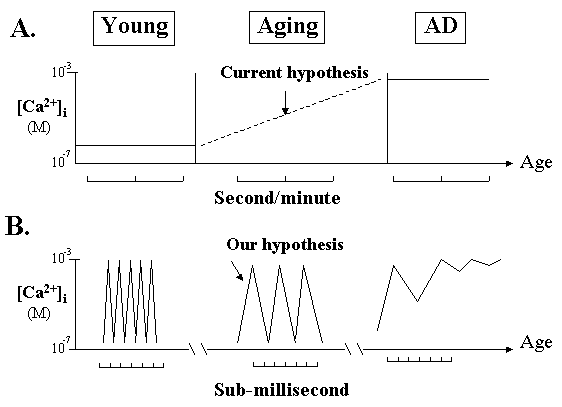
A. The current "calcium overload" hypothesis is based on the traditional concept of steady-state changes of Ca2+ measured at second or minute time intervals. It suggests that the average Ca2+ "levels" are steadily increasing during aging leading to cell death, therefore inhibiting Ca2+ entry will prevent AD.
B. Based on a Ca2+ oscillation concept, we propose that Ca2+ pulse frequency will progressively reduce during aging but this can be measured only at sub-millisecond time scale. If the frequency reduction surpasses a certain limit, Ca2+ gradient will collapse and cell will die. Thus, boosting the pulse frequency by Ca2+ agonists will slow down AD process. Note that the reduced pulse frequency means a "Ca2+ overstay", or "increased average Ca2+ levels", in the cytosol.
Fig. 2. Intermittent and alternate actions of the channels and pumps
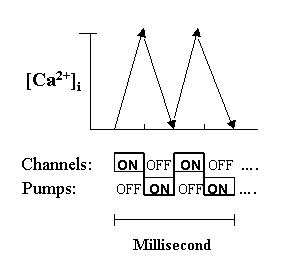
To elicit a net ascending slope, Ca2+ channels in a cell must be "on" at the same time while pumps must be "off". This is then followed by an immediate reversal to complete a spike within a fraction of a millisecond. Thus, channels and pumps must operate intermittently as an "integral unit" like an alternate generator. During aging, the turnover speed of the unit will slow down as a result of reduced energy input.
Fig. 3. Distinctive effects of Ca2+ agonists in the brain versus cultured cells.
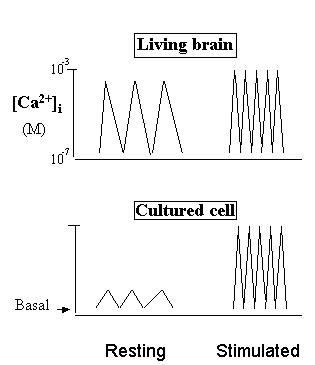
Why do Ca2+ agonists increase Ca2+ "levels" in the culture cells, but decrease them in the brain? In the living brain, Ca2+ is fully "potentiated" by action potentials and physiological Ca2+ agonists. But in the resting cultured cells, Ca2+ is pulsating at basal-line or surviving levels because the natural action potentials are absent. Therefore, addition of Ca2+ agonists (Stimulated) in the brain will reduce the time of Ca2+ stay in the cytosol (thus decrease Ca2+ "levels"), but increase the average Ca2+ "levels" in cultured cells if measured at second or minute time scale.
The Following Three Figures and a Table Summarize our "Natural Origin" Hypothesis for Plaques and Tangles
Updated -- 10 October 2001
Fig. 4. What will happen during aging?
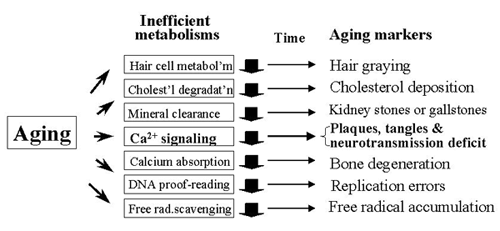
During aging, many biochemical pathways will naturally slow down, and Ca2+ signaling is no exception. Therefore, numerous aging markers, including plaques and tangles, will appear in essentially everyone and at about the same time in life.
Fig. 5. How have plaques and tangles formed during aging?
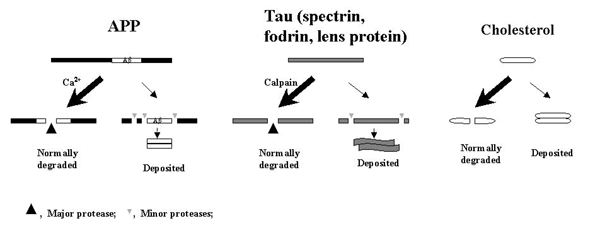
Proteins and other large molecules in our body will eventually end up in one of the two mutually exclusive outcomes: either degraded or deposited. If the deposition of cholesterol in aging is due to its insufficient normal degradation/clearance rather than "overactivation" of some abnormal factors, then a similar mechanism will underlie the depositions of APP and tau (and other calpain substrates).
Fig. 6. Subcellular localization of α-, β- and γ-secretases.
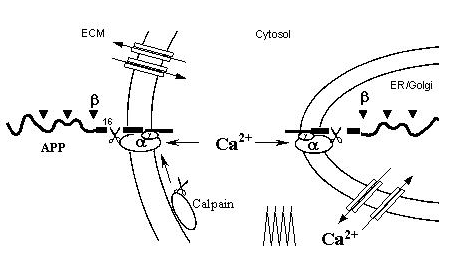
α-Secretase is most likely a Ca2+-dependent protease, perhaps calpain, because calpain is known to be active both in the cytosol and at cell surface. Such a "double membrane anchorage" of both a-secretase and APP would allow APP to be normally cleaved only at the lys-16 site. When Ca2+/calpain activity declines during aging, APP will accumulate and be progressively attacked by other proteases including β- and γ-secretases, until they reach the Ab core. (Note that calpain activity is regulated by Ca2+ spike frequency)
Table 1. Why is α-secretase most likely a Ca2+-dependent protease?
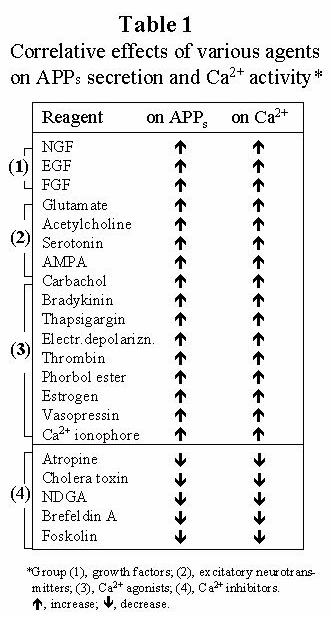
Because assuming a-secretase to be a Ca2+-dependent protease can explain the reported actions of these 21 agents altogether, and also because of the simplicity and explanatory power of the assumption for other AD features. Note that none of the other currently proposed candidates for α-secretase has been supported by comparable extensiveness and consistency of the data. Sources are listed in ref. 1. APPs, normally secreted APP.
Reference
1. Chen M. Alzheimer's a-secretase may be a calcium-dependent protease. FEBS Lett 1997; 417, 163-7. Abstract.
References
Other Citations
External Citations
Further Reading
No Available Further Reading
Comments
�
I have to admit that the emphasis
in calcium signaling can be a useful way to solve the
old controversy over the role of calcium in AD. Apparently,
experimentalists have been assuming that the time scale
of calcium signaling is the same as that of the instruments
they use to measure it. I agree, that does not have to
be the case. Calcium signaling is a relatively recently
studied phenomenon. All the mechanism we had at hand were
based in fluctuations of swimming-pool-like levels. However,
it imposes a technical problem to experimentalists and
Dr. Chen is not specific about the possible solutions.
�
Reply to Dr. Scatty by Ming Chen
Conceptualization of Ca2+ signals in the brain as dynamic "spikes" (1-3), instead of traditionally perceived steady-state "level" changes, is essential to explain some key dilemmas that have severely hampered our research progress. For example, growth factors promote cell growth by increasing Ca2+ levels, but cell death is also associated with increased Ca2+ levels. What, then, is the difference between these two seemingly the same but functionally life-or-death different "increased levels"? The "spike" concept now can distinguish them: The former is the increase of the spike FM and AM, but the latter is the collapse of the Ca2+ gradient (4).
The "spike" nature of Ca2+ (in and out cyclically) also suggests that channels and pumps in vivo would operate in a fascinating manner: They open/close INTERMITTENTLY and ALTERNATELY in a highly synchronized manner. This would be reminiscent of the way in which an alternate generator works - driven by energy and produces electric waves by rapidly changing the current directions. Evidently, when energy supply declines during aging, the turnover speed of the generator (i.e., the wave FM and AM) must REDUCE (action potentials and extrusion pumps are both driven by energy)(4).
Can this model be experimentally verified? While direct measurement of rapid Ca2+ spikes in the human brain has not been possible today, similar spikes have been elegantly measured first by Llinás and his colleagues in the giant nerves of LIVING squid in the sub-millisecond time scale (5, 6). (Note the dynamic Ca2+ spikes in vivo are largely driven by action potentials which are absent in the isolated cells; so only such measurements can reveal the GLOBAL Ca2+ states in the LIVING animal)(4).
Thus, it is reasonable to anticipate that the proposed age-related changes in spike frequency may also be directly measured in the OLD squid by current instruments. The study may also show us how the shape of the Ca2+ curve would look like when it is measured in a series of incremental time scales.
However, even if this is a success in squid, it still cannot be done in the human brain today; so how can we know what is happening in AD? A hypothesis can be directly verified by experimentation in some cases; but in others where current instruments are not up to the task, it may also be judged largely by the number of observations it explains. In this regard, our model is consistent with the fact that cognitive decline progresses as aging advances. It can also explain why growth factors and hormones have neuroprotective effects (they increase the spike frequency, not static "levels")(1-3). In contrast, current "Ca2+ rise" hypothesis may not: if Ca2+ levels are raised in aging cells, then why can growth factors, which should further increase Ca2+ entry thus kill cells, but have PROTECTIVE effects in reality?
Hypotheses developed on the available data need to evolve in accordance with new data and emerging concepts (1-3). This is particularly important when it comes to brain function of which we know so little (unlike AIDS or pneumonia where pathogenic causes can be directly seen).
However, this does not mean that we will not understand AD with current instruments. For example, we have known a lot about atom structure today even though nobody has actually seen it. We also know a lot about our origins even though nobody has directly seen that monkey becomes man. This means that deductive reasoning based on limited experimental data is of critical importance (perhaps more so than in other diseases) and may allow us to understand a significant part of AD, perhaps enough for designing a rational intervention strategy.
See also:
Kandel, ER et al. Principles of Neural Science. 3rd edn. (1991) Simon & Schuester, New York, p. 197
References:
Woods NM, Cuthbertson KS, Cobbold PH. Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature. 1986 Feb 13-19;319(6054):600-2. PubMed.
Berridge MJ. The AM and FM of calcium signalling. Nature. 1997 Apr 24;386(6627):759-60. PubMed.
Putney JW. Calcium signaling: up, down, up, down...what's the point?. Science. 1998 Jan 9;279(5348):191-2. PubMed.
Chen M, Fernandez HL. Ca2+ signaling down-regulation in ageing and Alzheimer's disease: why is Ca2+ so difficult to measure?. Cell Calcium. 1999 Sep-Oct;26(3-4):149-54. PubMed.
Llinás R, Steinberg IZ, Walton K. Presynaptic calcium currents in squid giant synapse. Biophys J. 1981 Mar;33(3):289-321. PubMed.
�
To my knowledge, there are probably over a hundred of reports showing that
calcium channel blockers have protective effects in old cells or animals and
even in AD patients. So, whatever your theory is (though it is interesting),
a rule is that one cannot argue against the facts. I think that these studies
have reasonably established that calcium, be it "levels" or "spikes", has overly
entered cells during aging and, therefore, blocking calcium channels will slow
down AD progression. This key discrepancy needs to be explained.
�
Reply to Dr. Aldoso by Ming Chen
In the world of research, we develop hypotheses based on facts. But perhaps the most difficult job is to judge, among the numerous reports, which ones are facts and which are not. Many papers have reported that calcium antagonists help old animals and AD patients. But, there are perhaps even more reports showing that growth factors and hormones protect aging cells. The latter agents exert biological effects by stimulating Ca2+ signaling, so they are physiological Ca2+ AGONISTS. More directly, two more recent statistic studies by Maxwell et al. (1) and Heckbert et al. (2) have examined 10,263 and 1,268 elderly subjects, respectively, and found that the frequent use of calcium channel blockers INCREASES the risk of AD-like symptoms in the elderly.
Now, which one to believe among these contradictory reports? What I do in this case is to see which of them fits in with the general biological principles.
First, Ca2+ channeling is essential for life and cognition (like ATP genesis and cAMP signaling) and, like any other basic metabolic pathways, it will deteriorate when life itself is approaching an end at advanced age. So, why do we want to further block it in the elderly?
Second, the Ca2+ “overload” hypothesis is based on a central assumption that Ca2+ channels are “overly opened” during aging. However, we know that this, if it happens, will require at least one of these two conditions: excess Ca2+ agonists (for ligand-gated channels) or excess energy (for voltage-gated channels). Evidently, neither of these can commonly occur during aging. So it is difficult to imagine that Ca2+ can excessively enter the cells during aging. In fact, if a functional Ca2+ over-influx occurs in humans, it should occur more likely in young adults (their energy and hormone/growth factor levels reach the peak), rather than in the elderly
Third, the Ca2+ overload hypothesis is originated in ischemia studies (where the dying cells exhibit Ca2+ rises). But several recent large-scale clinical trials on calcium antagonists have all failed in the ischemia patients (3). We know that Ca2+ extrusion pumps are highly energy-dependent (driving Ca2+ against a 10,000-fold gradient; no other metal has such a steep gradient). It follows that an acute energy crisis after ischemia will, among other things, slow down the pumps, resulting in a Ca2+ “overstay” in the cytosol (flattened spikes). Thus, what the damaged cells need is Ca2+ AGONISTS to boost their spike AM and FM, similar to the proposed strategy for rescuing the aging cells in AD (aging is accompanied by a mild and progressive energy shortage).
These considerations would perhaps cast reasonable doubts on the current theory and question the rationale for the LONG-TERM use of calcium channel blockers in the OLDEST-OLD people for AD prevention. (even though the drugs may give short-term relief in some common conditions such as hypertension which is related to vascular dementia, not typical AD).
The steady-state Ca2+ concept is derived from the observations that the usually measured average Ca2+ levels do correlate with physiological activities in CULTURED cells (grow, differentiation, etc.). So, this concept is not wrong, but like many other scientific concepts, has its limitation. For example, it does not explain the fact that Ca2+ agonists protect the aging BRAIN. So based on the cyclic nature of Ca2+ signals, we proposed a “Ca2+ spike generator” model.
Interestingly, the Ca2+ spike concept will also challenge some basic views today. For example, we take it for granted that Ca2+ agonists “open Ca2+ channels”. But if this is their only action, then how can the agonists increase the spike FREQUENCY? This clearly indicates that the agonists accelerate not only Ca2+ entry, but also its EXTRUSION (after each transient entry) during their actions.
But, how can the channel agonists accelerate extrusion by PUMPS that are driving Ca2+ in the OPPOSITE direction? And yet, what has activated the pumps when ONLY channel agonists are added to the cells?
These dilemmas would suggest to us that Ca2+ channels and pumps in vivo are functioning as an INTEGRAL WHOLE, which cannot be separated in concept. In other words, although the “Ca2+ spike generator” is made of channels and pumps, these “built-in” parts cannot be individually changed once the generator is in motion (life). As such, agents will not affect channels ONLY, but rather, they will either ACCELERATE or DECELERATE the turnover speed of BOTH channels and pumps as a whole. Indeed, accelerating the generator speed seems to be the exact action of the Ca2+ agonists in aging cells.
By the same token, calcium channel blockers will not block the channels only as it is widely thought. Rather, they will reduce the FM and AM of the Ca2+ spikes altogether. Thus, while it will take a long time for us to understand the precise mechanisms of Ca2+ signaling, but when it comes to the intervention strategies for AD, the issue should be black and white.
References:
Maxwell CJ, Hogan DB, Ebly EM. Calcium-channel blockers and cognitive function in elderly people: results from the Canadian Study of Health and Aging. CMAJ. 1999 Sep 7;161(5):501-6. PubMed.
Heckbert SR, Longstreth WT, Psaty BM, Murros KE, Smith NL, Newman AB, Williamson JD, Bernick C, Furberg CD. The association of antihypertensive agents with MRI white matter findings and with Modified Mini-Mental State Examination in older adults. J Am Geriatr Soc. 1997 Dec;45(12):1423-33. PubMed.
Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999 Jun 24;399(6738 Suppl):A7-14. PubMed.
�
A very stimulating hypothesis. But, a problem is that it is all based
on your findings in a single experimental paper suggesting that alpha-secretase may be a calpain. At the same time, however, there are over 50 or so proteases being also proposed today to be this or
other (beta- or gamma-) secretases, and some of them are supported by
many more data than yours (e.g., TACE or ADAM-10 as alpha-secretase). So I wonder how can you be so sure at this point that calpain is a better candidate than these other proteases for
alpha-secretase? Furthermore, beta-secretase has been positively
identified today and
maybe gamma- as well. Are they not the more direct intervention targets
than alpha-secretase to reduce Abeta in AD?
�
Reply to Dr. Wong by Ming Chen
First, let me take this opportunity to thank Drs. Wong, Scatty and Aldoso for raising in-depth and sharp questions. In science, asking the right questions sometimes can be as important as solving them. The AD field today needs a thorough debate on such long-standing issues.
Identification of APP secretases is fundamental in AD research but is extremely difficult. This is mainly because to claim a protease as a "secretase" implies that the protease is the ONLY or PRIMARY enzyme responsible for a specific cleavage in vivo (e.g., the lys-16 site). Because the cleavage specificity of most proteases is not restricted and many proteases can cleave the same site of a protein, this claim requires requires a demonstration that not only can a given protease cleave a certain site, but that NO OTHER PROTEASE can do the same. Evidently, one cannot make such a formidable claim unless ALL relevant proteases have been exhaustively studied and ruled out ONE BY ONE.
For this reason alone, we may question some of the current claims that certain secretases have been "positively identified." Because these studies have not excluded at least other currently proposed candidates for the same secretases, they remain CONTRADICTORY to one another (e.g., TACE, ADAM-10 and other proposed candidates for alpha-secretase, or BACE and many other proteases for beta- or gamma-secretases, respectively).
However, there is one unique protease whose activity on certain substrates may allow us to identify it definitively. This protease is calpain(s). It is a well-established criterion in the calpain study field that when filamin, talin, spectrin, tau, fodrin, and lens protein (in cataracts) are initially cleaved in intact cells or when cells are lysed by detergent, it is a definitive indication that calpain, and ONLY CALPAIN, is involved (1, 2). This unusual claim can be made mainly because the cleavage can be inhibited by Ca2+ chelator EGTA (or calpain inhibitors) in intact cells, and totally blocked by the same agents in cell lysates where an ENTIRE REPOSITORY of cellular proteases is present. This indicates that NO OTHER protease is involved in the same cleavage. Evidently, only a REGULATED protease acting on certain substrates has this advantage. Notably, Ca2+-dependent proteases are the only proteases that are known today to be dynamically REGULATED (though many proteases can be AFFECTED by various factors). I hope that these points can draw the attention of the investigators.
Now, we have accidentally observed that APP is cleaved at or near the lys-16 site under the same conditions where filamin and talin are cleaved and that APP remains intact when Ca2+/calpain is blocked (3). While this finding is subject to confirmation and different interpretations, it simply reminds us of the possibility that APP normal processing could be a Ca2+-regulated process, or APP could be a new member in the long list of physiological substrates of calpain.
This finding shocked us at first because it came at a time when an opposite notion had prevailed, i.e., "A-beta genesis is a Ca2+-regulated process" (see ref. 4). This is not an average discrepancy but is about Ca2+, a CENTRAL REGULATOR in cognition (which means that no other factor is more important than Ca2+ in cognition!). The current notion fits in perfectly with the "Ca2+ overload" dogma (5) which in turn dictates that "inhibiting Ca2+" would be a standard approach for AD intervention. Under this circumstance, our finding, which points to an opposite intervention strategy, would be unthinkable.
So, were we simply wrong? This dilemma haunted me for a long time until an idea emerged: APP processing has been vigorously studied and numerous agents have been reported to enhance or suppress alpha-processing. Thus, if the process is regulated by Ca2+ as we found, then it must follow that these agents should also activate or inhibit Ca2+ respectively as one of their cellular actions. In other words, these reports will serve as an independent judge for the two opposing views.
As I found in a careful analysis of numerous reports, the answer is clear-cut (4). These reports are made by many laboratories in a wide variety of cell types but exhibit an unusually high consistency with one another (for example, no study has shown that Ca2+ agonists can REDUCE APP normal secretion). Thus, while our experimental finding is a surprise, the corollary we deduced to it - "APP alpha-processing is Ca2+-regulated" - is deeply rooted. This claim is serious, and we welcome scholarly criticisms on any aspects of its experimental bases or reasoning steps. Here I take a brief look at some of the current challenges to it.
1. Can A-beta genesis be a "Ca2+-regulated" process? For one thing, Ca2+-regulated processes are ALL essential parts of life (cell growth, protein normal secretion, muscle contraction, neurotransmission, etc.) and their potencies are all REDUCING during aging. Because A-beta deposition is not essential part of life (it does not exist in young people) and it is INCREASING during aging, it cannot be a Ca2+-regulated process in concept. In fact, the general knowledge that APP alpha-processing correlates intimately with the activity of PKC, a Ca2+-regulated enzyme, clearly indicates that alpha-processing must be a Ca2+-related process. A recent report supports this view (6).
2. However, the intimate CORRELATION between alpha-processing and PKC activity has been widely interpreted as "alpha-secretase is REGULATED by PKC". But, if alpha-processing is Ca2+-regulated, then it should be expected to correlate with many Ca2+-dependent enzymes (they will all rise or fall with Ca2+)(7). Now, if a Ca2+-regulated PROTEASE or calpain can directly explain the CLEVAGE of APP after Ca2+ activation, then by Occam's razor, one should not increase, beyond what is necessary, the number of factors to explain anything.
3. Our data have not unambiguously established that calpain cleaves APP at exactly the lys-16 site and some sequencing studies have found that in addition to a major APPs (secreted APP), other slightly longer or shorter APPs species also exist in cells (see ref. 4). Based on these data it has been suggested that there should be several "alpha-like" secretases acting on different sites of APP (if so, then identifying one of them would be of little use).
It must be noted that numerous sequencing studies by CONVENTIONAL method (chemical sequencing) have shown that APPs from a wide variety of cell types all ends at lys-16 (see ref. 4). This indicates that a highly site-specific protease acting at lys-16 is predominant (this high site-specificity is ensured by a unique "double membrane anchorage" of both alpha-secretase and APP)(Fig. 1 in ref. 4). On the other hand, the multiple APPs species are found only by radio-labeled sequencing. This method is much more sensitive thus will detect many MINOR or TRACE species in addition to the MAJOR one. For this reason, the latter results do not confront, but only add-on to, the former. Even if several "alpha-like" proteases exist, should they be of the same importance? Cholesterol plaques, too, contain many other trace lipids, should the intervention in atherosclerosis, therefore, target all of them equally? Or simply the major one?
4. Alpha-secretase is up-regulated in the activated cells, but resting cells also have a baseline APPs secretion which cannot be totally blocked. This fact seems to suggest that there are "two alpha-secretases" acting on the same APP site: one regulated, the other unregulated (8).
There may be an alternative explanation for the fact. Protein secretion is ESSENTIAL for life, so resting cells must have a baseline or survival level of protein secretion because they are ALIVE; and totally blocking the secretion would mean cell death. One might compare this situation to other essential pathways which also cannot be totally blocked in the resting cells, such as ATP synthesis, albumin secretion, and DNA replication. If resting cells have these processes in baseline levels, and cell activation stimulates the same rather than "new" pathways, then it would be reasonable to conceive that the cell manipulations have simply affected the same alpha-secretase, but at different FUNCTIONAL STATES.
5. Gene mutations, head trauma, ischemic injury, and many other pathological (erroneous) processes are known to increase A-beta genesis (see ref. 7). Based on these facts it has been assumed that similar errors/insults may be responsible for plaque formation in most AD patients. This reasoning has boosted an exhaustive search of numerous metabolic "mistakes" believed to underlie amyloid plaque formation. But is the reasoning correct?
Rare gene mutations (e.g., on LDL receptor genes) can cause cholesterol deposition in juvenile/familial atherosclerosis, but we know that the "same" cholesterol deposition in most elderly is not due to gene mutations, but a result of AGING. Although many abnormal pathways can lead to A-beta overproduction, it should be clear that there is ONLY ONE reason underlying plaque formation in most elderly, and this reason is NORMAL AGING (7).
Why normal aging can lead to amyloid plaques? Among the numerous possible models, we again prefer the simplest one. During aging, the activity of Ca2+/alpha-secretase will decline so some intact APP will accumulate. This will allow other proteases to attack it. These attacks will continue until they reach the core of APP (A-beta) which is aggregating and resistant to further attacks. So, in the end, APP will become uniform A-beta aggregates and remains as such until after the person's death. Thus, postmortem examination of the brain will find mainly the uniform A-beta. In this context, plaques would be a NATURAL remnant of aging, like crud in the old plumbing, a result of insufficient NORMAL degradation/clearance, but not due to any "abnormal" pathways, similar to the mechanisms for cholesterol or gallstone depositions during aging (details in ref. 7).
6. However, an overwhelming theory today is that "abnormal" beta- and gamma-secretases are "overactivated" and they have "specifically" cleaved A-beta out of APP before alpha-secretase can act. By this model, cholesterol and gallstone depositions would also be due to "overactivated" cholesterol "deposit-ases" and mineral "precipit-ases". Readers can judge for themselves which model is more reasonable.
As mentioned above, once calpain is inactivated, other proteases usually will not attack the calpain substrates at the same site (otherwise they would not be protected by EGTA alone in cell lysates). This unique feature allows spectrin, tau, fodrin, and lens protein to accumulate together with APP during aging. More accurately, it is their calpain normally-attacking cores that are accumulated while other portions of the proteins are degraded, similar to the A-beta core deposition (filamin and talin are in the peripheral cells which are rapidly turning over thus not deposited). Unfortunately, these depositions are widely viewed today as the results of "calpain overactivation".
Notably, our model can also explain why presenilin and APP gene mutations can cause the same amyloid deposition (they both reduce APP normal degradation: the former reduces Ca2+/alpha-secretase activity, the latter changes APP structure)(4, 7). It can also explain tangle formation (7). This mechanism, in essence, coincides with a recent report that the deposition of alpha-synuclein in Parkinson's is due to INSUFFICIENT normal degradation of the protein (9).
7. It has also been proposed that amyloid plaques may be formed by abnormal aggregation, fibrillization or mis-folding of A-beta protein, implying that there is an unidentified "abnormal" factor which triggers the soluble A-beta to aggregate in AD (10).
If this is the case, then why can A-beta and many other proteins aggregate and fibrillize SPONTANEOUSLY in test tube at sufficient concentrations? It clearly indicates that aggregation and fibrillization are NATURAL attributes of many proteins at certain concentration threshold. This threshold will be crossed when proteins start to accumulate during aging.
Many controversies today can be traced to the way in which the initial question was asked, i.e., "how are plaques and tangles formed in AD"? Because AD is an abnormal brain function, the answer would be naturally the abnormal factors. However, had it been asked "how are they formed during NORMAL AGING?", then the answer may have been quite different. Although plaques and tangles deposit at a faster rate and thus are more abundant in AD patients than in the age-matched controls, this can be generally explained by RISK FACTORS in life (exercise, diet, individual background, etc), again similar to the faster depositions of cholesterol and gallstones in some people. As such, the best way to slow down their accumulation is to boost the inefficient NORMAL pathways, thus fundamentally different from the strategies for treating typical PATHOGENIC processes such as HIV proliferation or cancer growth. It also implies that AD needs to be explained by factors more profound than plaques and tangles.
References:
Chen M, Stracher A. In situ phosphorylation of platelet actin-binding protein by cAMP-dependent protein kinase stabilizes it against proteolysis by calpain. J Biol Chem. 1989 Aug 25;264(24):14282-9. PubMed.
Croall DE, DeMartino GN. Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiol Rev. 1991 Jul;71(3):813-47. PubMed.
Chen M, Durr J, Fernandez HL. Possible role of calpain in normal processing of beta-amyloid precursor protein in human platelets. Biochem Biophys Res Commun. 2000 Jun 24;273(1):170-5. PubMed.
Chen M. Alzheimer's alpha-secretase may be a calcium-dependent protease. FEBS Lett. 1997 Nov 10;417(2):163-7. PubMed.
Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann N Y Acad Sci. 1994 Dec 15;747:1-11. PubMed.
Skovronsky DM, Lee VM, Praticò D. Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J Biol Chem. 2001 May 18;276(20):17036-43. PubMed.
Chen M, Fernandez HL. Where do Alzheimer's plaques and tangles come from? Aging-induced protein degradation inefficiency. Front Biosci. 2001 Mar 1;6:E1-E11. PubMed.
Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998 Oct 23;273(43):27765-7. PubMed.
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science. 2001 Jul 13;293(5528):263-9. PubMed.
Jarrett JT, Lansbury PT. Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie?. Cell. 1993 Jun 18;73(6):1055-8. PubMed.
�
This debate makes me rethink some basic issues in AD. Here I have another question.
Calcium is important for brain function, no problem.
But many other factors are also important such as aluminum,
zinc, iron, acetylcholine, mitochondria, cAMP,or NO.
Brain will not work without any one of them, and imbalance
in these factors has caused brain disorders.If so, then
how can calcium be singled out as a central regulator
in cognition and no other factor is more important than
calcium in cognition? Furthermore, if plaques and tangles
are "normal" products of aging, then what on earth has
caused horrible AD?
�
Reply to Dr. Wong by Ming Chen
Many factors are required in brain function, but their roles are not EQUAL. Ca2+ is the only factor that is known today to be both NECESSARY and SUFFICIENT for triggering and controlling neurotransmitter release (any textbooks). This means: no Ca2+, no neurotransmission; and once Ca2+ is there, nothing else is needed (in intact cells). Such a role of Ca2+ to neurotransmission would be like a key is to a lock. So, any factors, if they affect neurotransmission (such as aging, gene mutations, head injury, exercises, or diets), should most likely do so by directly or indirectly but eventually affecting Ca2+ (like everyone can open the lock, but perhaps no one can do so without using the key).
Such a central role of Ca2+ in cognition may be comparable to insulin in glucose metabolisms. Although many factors can contribute to diabetes (i.e., reduced glucose catabolism), replenishing insulin can improve conditions in many patients. This suggests that those factors act by converging their actions eventually on the insulin signaling system (either causing insulin deficiency or the system dysfunction). This would make me think that studying AD without focusing on Ca2+ signaling systems (which includes many related systems and factors) would be like studying diabetes without focusing on insulin.
Why is Ca2+ so unique in the brain? Probably, we can compare the brain to a symphony orchestra, which must have a conductor. But who can take this central position? Certainly not everybody, because the first requirement for a conductor is that he/she must carry a large body of instructions for coordinating ALL instruments. Now, if Ca2+, a simple metal, is a central regulator in the brain, then how can it carry enough information in controlling the countless chemical reactions in cognition?
Its "spike" nature may suggest a clue. The FM and AM changes of Ca2+ spikes can give rise to INFINITE combinations of wave shapes, thus can carry with them an amount of information that is virtually UNLIMITED, like radio waves can transmit any TV pictures. During cognition, the "digital" information encoded in the Ca2+ spikes is decoded by many downstream Ca2+-regulated processes, especially neurotransmitter release through its variations (type, combination, intensity, interval and duration, etc.) - in much the same way as the conductor's instructions are decoded by the instrument players (1).
On the other hand, no other factor is known today to have a comparable information-carrying capacity, not even close. For example, cAMP, cGMP, NO, or any other metals (Na+ and K+ pulses in action potentials induce transmitter release only through Ca2+ spikes; DNA carries enormous information for the brain structures, but perhaps not for higher cognition, an acquired ability). This consideration suggests that Ca2+ signaling, by necessity, will stand at a CENTRAL position in the hierarchical orders of the numerous factors in the brain.
However, even if Ca2+ is most important in cognition, that does not mean that cognitive disorders are always due to Ca2+ defects. In fact, any instrument failure in the orchestra will disrupt the performance. So why do we think that Ca2+ deficit is an initial defect in AD?
This model does not come from a pure imagination, but is the outcome of our systematic analysis of the many unique features of the disease. For example, although imbalance in aluminum, iron, vascular disease, or HIV can all cause dementia, these types of dementia nevertheless differ from AD in one critical feature: only AD is accompanied by a chronic accumulation of plaques and tangles. So, any models intended to explain AD also need to explain the origins of the hallmark lesions.
Now, if APP normal processing is Ca2+-related (simply because it correlates with PKC activity), tau is normally degraded by calpain and de-phosphorylated by phosphatases including calcineurin (well-known), and Ca2+ signaling itself will decline during aging (see above), then what would you think of the possible origin of AD?
Further, if plaques and tangles are accumulated at a faster rate in AD patients than in other elderly, and so is the rate of neurotransmission decline than other functions in the patient's body, then what would you think of the primary target for AD intervention?
A common assumption today is that it is either plaques or tangles that cause AD. If they play such a classic "cause-effect" role, then plaques or tangles would act like conventional pathogens such as HIV, arsenic or mercury. But if this is the case, then we would face a more difficult dilemma: While classic pathogens kill everybody they invade, why can most elderly remain perfectly healthy after having carried plaques and tangles in the brain for decades?
Yet, if those inert protein remnants caused AD, then why can the AD process be so sensitively influenced by such subtle factors as education, social contacts, and even story-telling (see AD website)? This puzzle, however, may be explained if Ca2+ is assumed to be the initial defect because it is most sensitively and dynamically regulated by a myriad of factors.
Very recently, an interesting progress is that several authors, who had previously reported a "Ca2+ overflow" into the cytosol of AD cells, but now found that Ca2+ has "overfilled" the Ca2+ stores such as ER (2, 3). Notably, cytosol is the only place where Ca2+ spikes take place, so it is like the combustion chamber of an engine where life is, but ER is like a gas tank. Now, if the tank is overfilled in AD cells, then will the chamber have sufficient gas to use?
Regardless of the precise mechanisms of Ca2+ signaling, today both Ca2+ antagonists (e.g., nimodipine) and agonists (e.g., estrogen) are being prescribed to MILLIONS of elderly each year. Thus, if Ca2+ is important and somehow altered in aging as we all agreed on, then it must follow that one of the drugs would increase the AD risk in the elderly perhaps by the same scale. Can any other questions in AD research be more important and urgent than this one?
However, this question may not be fully resolved without addressing several difficult issues at the same time: If Ca2+ deficit, plaques and tangles are all normal products of aging as we proposed, then what has caused the horrible, ABNORMAL brain function in some elderly, but not in others? More puzzling, the issues discussed here are mostly nothing but common-sense knowledge, how can they become the subjects of debate in a field that has been going on for decades? Yet, the initial causes of many human diseases have been largely understood after only a few years of research, but why has late-onset sporadic AD remained so enigmatic after perhaps the most vigorous crusade for so many years?
These questions are much more profound and may need a bottom-up examination of several basic concepts that have been guiding our research including the definition of the disease. But this is beyond the scope of the current debate (which focuses on the origin of plaques and tangles themselves and Ca2+ changes). But those questions must be addressed in AD research. Hence, they will be the subjects in another debate which will start soon.
References:
Putney JW. Calcium signaling: up, down, up, down...what's the point?. Science. 1998 Jan 9;279(5348):191-2. PubMed.
Mattson MP, Chan SL, Camandola S. Presenilin mutations and calcium signaling defects in the nervous and immune systems. Bioessays. 2001 Aug;23(8):733-44. PubMed.
Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, Laferla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000 May 15;149(4):793-8. PubMed.
Make a Comment
To make a comment you must login or register.