Methyl Groups Put the Brakes on FTD Progression
Quick Links
In some people who carry a repeat expansion in the C9ORF72 gene, methylation of the gene promoter shuts down its transcription. This offers a modicum of protection against amyotrophic lateral sclerosis and frontotemporal dementia, according to a paper in the March 20 Neurology online. Although the silencing did not stave off disease, it seemed to slow brain atrophy and memory decline in a small cohort studied at the University of Pennsylvania Perelman School of Medicine in Philadelphia. However, experts agree that more work is needed to build consensus that methylation of C9ORF72 definitely protects the brain.
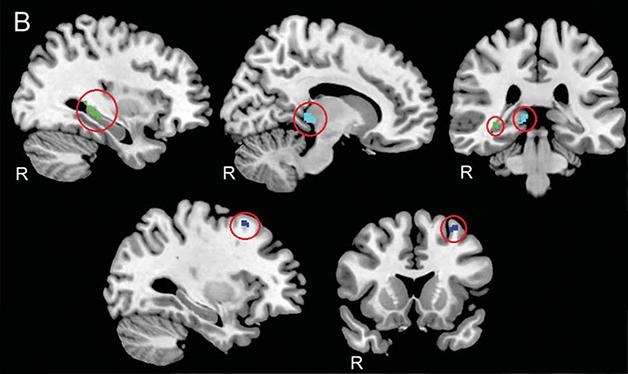
Safe Zones. C9ORF72 methylation protected three regions of the brain (circled): the hippocampus (green), thalamus (cyan), and premotor cortex (blue). [Republished with permission, © 2015 American Academy of Neurology.]
Neuroscientists do not know what C9ORF72 does, and struggle to reconcile dueling proposed mechanisms for disease. One is that the hexanucleotide repeats interfere with the protein’s normal function, causing neurodegeneration by haploinsufficiency. The alternative is that the repeats, known to produce abnormal RNA and peptide products, instigate disease due to a toxic gain of function (see Jan 2013 news; Feb 2013 news; news; Oct 2013 news).
Senior author Edward Lee suspected both theories had merit, but wanted to know which mechanism might dominate. His group and Ekaterina Rogaeva’s at the University of Toronto had found that methyl groups decorated the C9ORF72 promoter in some people with the expansion. This offered an opportunity to answer his question, because methylation typically silences genes (see Jun 2013 news; Liu et al., 2014; Xi et al., 2014). If haploinsufficiency was the main culprit, then methylation should make the disease worse. If gain of function was more important, then methylation ought to help by shutting off transcription into aberrant RNAs encoding undesirable repeat peptides.
In a study earlier this year, Lee and colleagues reported that methylation of the C9ORF72 promoter did not affect whether carriers developed ALS or FTD or when they got it, but it did correlate with slower progression in people who had only FTD (Russ et al., 2015). However, this result was cross-sectional and retrospective, based on patient charts and autopsies. Lee wanted to observe the methyl groups’ effects in a longitudinal study of living people. He approached first author Corey McMillan, who was performing magnetic resonance scans and neuropsychological tests on expansion carriers. Together they studied a mixed group of 20 patients, most with behavioral variant FTD, but some with primary progressive aphasia, another FTD subtype, ALS-FTD, or ALS with mild cognitive impairment. All carried repeat expansions, though the researchers did not quantify how long those were.
Lee assessed methylation status via DNA extracted from blood samples, which tends to have the same methylation pattern as DNA in the brain, researchers say. C9ORF72’s promoter contains 26 CpG nucleotide dyads that are potential methylation sites. Lee checked just one, which he said typically correlates with methylation elsewhere in the region. However, even within a blood sample, C9ORF72 is more methylated in some cells than others, so the authors aimed to quantify what proportion of cells had methylated the promoter. Lee treated the DNA samples with a restriction enzyme that cuts only unmethylated DNA. Then he used polymerase chain reaction to amplify that specific methylation site. If the blood cell population contained mostly unmethylated DNA, then the PCR would take many cycles to produce products, because much of the target DNA would be chopped up. Conversely, in a heavily methylated sample, the PCR would work quickly. Lee found that the DNA samples contained anywhere from zero to 30 percent methylation; that is, up to 30 percent of the cells in a blood sample had methylated the C9ORF72 promoter.
Next, the authors examined gray-matter density in MRI scans of the 20 patients. Compared with healthy controls, the expansion carriers exhibited widespread reductions in gray-matter density, including in the frontotemporal cortex and cerebellum. When the authors considered methylation status and gray matter together, three regions stood out. The more methylation a person had, the denser the gray matter in his or her hippocampus, premotor cortex, and thalamus. Repeat scans were available for 11 of the study participants, allowing the researchers to assess change over time. They report that the same regions atrophied more slowly in people with heavily methylated C9ORF72 promoters, suggesting that methylation was protective in those particular spots. From previous studies, these regions were known to be particularly vulnerable to C9ORF72-based FTD compared with sporadic disease (Bede et al., 2013; Irwin et al., 2013).
McMillan also tested 10 subjects for cognitive decline. The ability to recall a word list, a task that taxes the hippocampus, fell more sharply in volunteers with relatively unmethylated C9ORF72. Finally, examining autopsy tissue from 35 people, Lee connected higher levels of methylation to a greater number of neurons in the hippocampus and frontal cortex than in those with a lower percentage of methylation (thalamus tissue was unavailable).
The authors conclude that methylation looks to be protective, though concede they have not proven it. Jeremy Day and Erik Roberson of the University of Alabama, Birmingham, who penned an editorial accompanying the study, agreed. “These findings indicate that epigenetic mechanisms counter genetics at C9ORF72, with DNA methylation suppressing effects of the repeat expansion,” they wrote. They added, “These data may spell the beginning of the end for the loss-of-function hypothesis of C9ORF72 pathogenesis.” Lee cautioned, however, that no one has found any effect of methylation on the onset of ALS or FTD. C9ORF72 haploinsufficiency could still kick off the disease, he speculated, with the production of toxic RNAs and peptides dominating the process later.
Indeed, methylation appeared to speed up the disease in Rogaeva’s study of 37 expansion carriers with ALS. Why the discrepancy? Both cohorts were small, making it possible that one or the other had a false positive result. In addition, one study focused on ALS, one on FTD. “The biggest weakness of this study was that people had varied diagnoses,” Lee added. If methylation influences what kind of FTD a person gets, then his cohort, with mostly behavioral variant FTD, might be skewed toward the level of methylation associated with that particular diagnosis. Then the speed of progression might have more to do with the kind of FTD, rather than the methylation status itself, he said. Rogaeva, who was not involved in the new study, also noted that McMillan was unable to assess spinal cord atrophy via MRI—the cord is too skinny to resolve that way—and it remains possible that the high-methyl patients had plenty of atrophy there.
Rogaeva found the work interesting, but said many questions must be answered before researchers can agree that methylation protects. For one, it would be necessary to assess methylation more completely and quantitatively. She was not convinced that looking at one or two CpG sites in the promoter constitutesd a solid indicator for methylation overall. Moreover, Rogaeva recently reported that methyl groups also decorate the repeats themselves, though she cannot yet quantify how methylated the expansion gets (see Mar 2015 news). She suggested that a thorough analysis of methylation in a larger, phenotypically homogeneous population might yield more convincing results. Lee, for his part, is experimenting with C9ORF72 promoter methylation in cell culture models, looking for results consistent with his human studies.
If Lee and McMillan are correct, then treatments that methylate C9ORF72, or otherwise silence it, could be promising. Scientists are already experimenting with antisense oligonucleotides to C9ORF72 (see Oct 2013 news; news).—Amber Dance.
References
News Citations
- Chicago—RNA Inclusions Offer Therapeutic Target in ALS
- RNA Twist: C9ORF72 Intron Expansion Makes Aggregating Protein
- Second Study Sees Intron in FTLD Gene Translated
- RNA Deposits Confer Toxicity in C9ORF72 ALS
- Methylation a Turn Off for Disease Gene C9ORF72?
- Expansion and Methylation Go Hand in Hand in C9ORF72
- Second Study Confirms Antisense Oligonucleotides Bust RNA Aggregates
Paper Citations
- Liu EY, Russ J, Wu K, Neal D, Suh E, McNally AG, Irwin DJ, Van Deerlin VM, Lee EB. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014 Oct;128(4):525-41. Epub 2014 May 8 PubMed.
- Xi Z, Rainero I, Rubino E, Pinessi L, Bruni AC, Maletta RG, Nacmias B, Sorbi S, Galimberti D, Surace EI, Zheng Y, Moreno D, Sato C, Liang Y, Zhou Y, Robertson J, Zinman L, Tartaglia MC, St George-Hyslop P, Rogaeva E. Hypermethylation of the CpG-island near the C9orf72 G₄C₂-repeat expansion in FTLD patients. Hum Mol Genet. 2014 Nov 1;23(21):5630-7. Epub 2014 Jun 6 PubMed.
- Russ J, Liu EY, Wu K, Neal D, Suh E, Irwin DJ, McMillan CT, Harms MB, Cairns NJ, Wood EM, Xie SX, Elman L, McCluskey L, Grossman M, Van Deerlin VM, Lee EB. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015 Jan;129(1):39-52. Epub 2014 Nov 12 PubMed.
- Bede P, Elamin M, Byrne S, McLaughlin RL, Kenna K, Vajda A, Pender N, Bradley DG, Hardiman O. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology. 2013 Dec 10;81(24):2107-15. Epub 2013 Nov 8 PubMed.
- Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J, Rascovsky K, Toledo JB, Boller A, Bekisz J, Chandrasekaran K, Wood EM, Shaw LM, Woo JH, Cook PA, Wolk DA, Arnold SE, Van Deerlin VM, McCluskey LF, Elman L, Lee VM, Trojanowski JQ, Grossman M. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013 Feb;84(2):163-9. PubMed.
Further Reading
Papers
- Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, Whitwell J, Foti D, McDade E, Molano J, Karydas A, Wojtas A, Goldman J, Mirsky J, Sengdy P, Dearmond S, Miller BL, Rademakers R. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011 Feb;82(2):196-203. PubMed.
- Mahoney CJ, Downey LE, Ridgway GR, Beck J, Clegg S, Blair M, Finnegan S, Leung KK, Yeatman T, Golden H, Mead S, Rohrer JD, Fox NC, Warren JD. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. 2012 Sep 24;4(5):41. PubMed.
- Belzil VV, Bauer PO, Gendron TF, Murray ME, Dickson D, Petrucelli L. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 2014 Feb 12; PubMed.
- Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, Dejesus-Hernandez M, Rutherford NJ, Baker M, Knopman DS, Wszolek ZK, Parisi JE, Dickson DW, Petersen RC, Rademakers R, Jack CR, Josephs KA. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012 Mar;135(Pt 3):794-806. PubMed.
- Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013 Dec;126(6):895-905. Epub 2013 Oct 29 PubMed.
News
- Brain Imaging Distinguishes C9ORF72 From Other Types of ALS
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
- Drug and Biomarker Candidates for C9ORF72 ALS and FTD
- C9ORF72’s Dirty Work Done by Problem Proteins
- C9ORF72 Killer Dipeptides Clog the Nucleolus
- RNA-DNA Pairs: At the Root of C9ORF72 Repeat Damage?
Primary Papers
- McMillan CT, Russ J, Wood EM, Irwin DJ, Grossman M, McCluskey L, Elman L, Van Deerlin V, Lee EB. C9orf72 promoter hypermethylation is neuroprotective: Neuroimaging and neuropathologic evidence. Neurology. 2015 Apr 21;84(16):1622-30. Epub 2015 Mar 20 PubMed.
- Day JJ, Roberson ED. DNA methylation slows effects of C9orf72 mutations: An epigenetic brake on genetic inheritance. Neurology. 2015 Apr 21;84(16):1616-7. Epub 2015 Mar 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Sheffield
This interesting study is particularly notable for the way it integrates methylation analysis with a number of other data types, including clinical symptoms, neuropathology, and neuroimaging. Although the study includes a relatively small number of patients, the authors' success contrasts with similar studies attempting to correlate C9ORF72 repeat length (as measured by Southern blotting) with disease severity and supports their methodology, but also suggests that future work should integrate region-specific analysis of C9ORF72 repeat length with methylation status. Overall this data is supportive of other work suggesting that C9ORF72 repeat expansions produce gain-of-function toxicity—hypermethylation-mediated transcriptional silencing, which reduces disease severity measured clinically, pathologically, or using imaging. In a previous study the authors have also observed correlation between hypermethylation and reduction in RNA foci and dipeptide repeat proteins derived from the repeat sequence.
View all comments by Johnathan Cooper-KnockMake a Comment
To make a comment you must login or register.