New Tracer Shows Disrupted Glutamate Dynamics Near Plaques
Quick Links
Amyloid plaques are thought to precipitate cognitive decline by creating a toxic microenvironment for neurons, but scientists are still working out the exact mechanism. Now, using a new, high-resolution in vivo imaging methodology, researchers led by Jasmin Hefendehl and Brian MacVicar at the University of British Columbia, Canada, report that glutamate transport falters on the fringes of plaques. It no longer removes the neurotransmitter from the synapse, causing aberrant glutamate signaling. By boosting glutamate transport pharmacologically, the authors were able to tamp down random glutamate fluctuations and return glutamate dynamics to normal in a mouse model of Alzheimer’s disease. The authors claim that this could be a therapeutic strategy. Hefendehl and colleagues reported these findings in the November 11 issue of Nature Communications.
“This is an important study with major implications for understanding the mechanisms of plaque-related hyperactivity and glutamate cytotoxicity in AD,” said Saak Ovsepian, Technical University of Munich, Germany. He was not involved in the work.
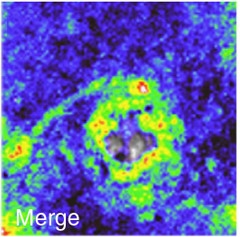
Glutamate Storm.
Around plaques (gray center), glutamate levels ramp up. [Courtesy of Jasmin Hefendehl, Nature Communications.]
To investigate glutamate dynamics in relation to plaques, Hefendehl availed herself of iGluSnFR—a new, highly sensitive glutamate marker engineered by fusing a variant of GFP to the bacterial glutamate binding protein GLT-I and coupling to a synapsin promoter for expression in neurons. This is the first time iGluSnFR has been used to investigate AD.
The authors injected AAV viruses coding iGluSnFR into either the somatosensory or visual cortices of five- to six-month-old wild-type and APPPS1 transgenic mice. At this age, APPPS1 mice have accumulated amyloid plaques in the brain, but perform normally in learning and memory tests. The authors aimed to look for cellular signaling changes that occur early in disease pathology.
Ten days after introducing iGluSnFR, the researchers implanted a clear window into the cranium above the injection site. A week later they placed the anesthetized mouse under a two-photon microscope and recorded iGluSnFR fluorescence before and after either shining a light in the mouse’s eye or tapping its leg. In wild-type mice both these sensory stimuli released a burst of synaptic glutamate.

Glutamate Gone Awry.
In APPPS1 transgenic mice, the glutamate response induced by a physiological stimulus is disrupted in regions of interest closest to amyloid plaque (ROI1) compared to those farther away (ROI4) . [Courtesy of Jasmin Hefendehl.]
In transgenic mice, by contrast, the stimuli did not register near plaques. In fact, even before the stimulus, glutamate levels fluctuated wildly and randomly close to these deposits (see image at right). A few dozen microns away, glutamate did spike in response to light or touch, but it dissipated more slowly than in wild-type mice. All told, the findings indicated that glutamate dynamics around plaques had gone gaga.
What explains these changes? Using immunohistochemistry, Hefendehl and colleagues found less endogenous mouse GLT-1 in the vicinity of plaques in APPPS1 transgenic mice. This could explain the high baseline activity and slow dissipation of glutamate signals, since this transporter primarily functions to rapidly clear glutamate from synapses.
To test whether these glutamate disruptions affect neurons, the authors switched to calcium imaging to measure neural activity at various distances from plaques. Fewer neurons close to plaques responded to the stimulus, but their average Ca2+ burst was the same regardless of location.
How is it that some neurons can be swimming in glutamate at baseline, have no stimulus-induced glutamate surge, and still fire as though they were perfectly healthy? Roberto Malinow, University of California San Diego, takes these preserved calcium signals as a lack of evidence for changes in neuronal behavior due to Aβ. Hefendehl thought otherwise. She reasoned that average Ca2+ bursts probably did not correlate with distance from plaques because even neurons close to plaque receive input from healthier tissue far from the glutamate-flooded zone.
Ovsepian told Alzforum the results were convincing and fit with prior studies linking glutamate toxicity to Aβ pathology. Earlier this year, Ovsepien and colleagues reported that extrasynaptic metabotropic glutamate receptor signaling in the vicinity of plaques might contribute to hyperactivity of pyramidal cells in the cortex of APPPS1 and APP23 mice, building on previous work linking Aβ toxicity to activation of these receptors (Ovsepian et al., 2016; Jun 2009 news; Jun 2010 news).
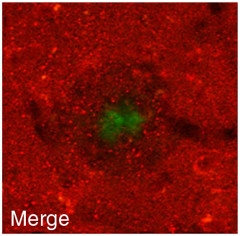
Glutamate Transporter.
Decreased GLT-1 (red) near amyloid plaque (green).
Curiously, Hefendehl and colleagues partially restored glutamate dynamics in the APPPS1 mice by giving them ceftriaxone, an antibiotic that also boosts expression of GLT-1. MacVicar said this was the most surprising finding. Hefendehl measured iGluSnFR fluorescence before and after five days of systemic ceftriaxone treatment. It reduced baseline fluctuations of glutamate near plaques and quickened the pace of glutamate clearance after a stimulus-induced burst. Neurons near the plaque still didn’t register the stimulus, so glutamate function recovered only partially. Nevertheless, if ceftriaxone performs similarly in the human brain, MacVicar said, it could open the door to new ways of treating AD. David Cook, University of Washington School of Medicine, cautioned that as a potent antibiotic, ceftriaxone probably won’t be useful in treating a chronic condition such as AD directly. MacVicar agrees but nevertheless thinks ceftriaxone could be tried as an experimental treatment. Ceftriaxone is FDA-approved. It has undergone a Phase 3 clinical trial for restoring muscle strength in more than 500 people with ALS, where it failed (see clinicaltrials.gov; Jan 2013 news). —Erin Hare.
Erin Hare is a freelance writer based in Pittsburgh.
References
Research Models Citations
News Citations
- Neuronal Glutamate Fuels Aβ-induced LTD
- Aβ Oligomers: A Fatal Attraction for Glutamate Receptors?
- Chicago—ALS Clinical Trials: New Hope After Phase 3 Setbacks
Paper Citations
- Ovsepian SV, Blazquez-Llorca L, Freitag SV, Rodrigues EF, Herms J. Ambient Glutamate Promotes Paroxysmal Hyperactivity in Cortical Pyramidal Neurons at Amyloid Plaques via Presynaptic mGluR1 Receptors. Cereb Cortex. 2016 Sep 6; PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Hefendehl JK, LeDue J, Ko RW, Mahler J, Murphy TH, MacVicar BA. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat Commun. 2016 Nov 11;7:13441. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Boston University Chobanian & Avedisian School of Medicine
These findings, along with those of McEwen and colleagues looking at riluzole, and those of Duff and colleagues looking at the role of hyperactivity in the spread of tau neuropathology in AD models, provide compelling data in support of additional preclinical studies looking at pharmacological modulation of excitatory neurotransmission as a potential therapeutic target in AD, especially if such therapeutic interventions can be initiated before the onset of overt clinical manifestations.
Make a Comment
To make a comment you must login or register.