Do APP and Parkinson’s Proteins Collude to Cause Neurodegeneration?
Quick Links
Two recent studies are charging that there is collusion between Parkinson’s disease proteins and the amyloid precursor protein (APP), in particular its proteolytic progeny, the amyloid intracellular domain (AICD). In the first paper, published July 18 in Science Signaling, Li Zeng and Eng-King Tan of the National Neuroscience Institute in Singapore report that the PD-associated kinase LRRK2 phosphorylates APP at a site within the AICD, and that this promotes AICD’s nuclear localization and neurotoxicity. The researchers’ linkage of LRRK2 to APP processing and toxicity drew some critique from outside commentators. In a second report in the May 3 Biological Psychiatry, Cristine Alves da Costa and Frederic Checler of the Institut de Pharmacologie Moléculaire et Cellulaire in Valbonne, France, tie AICD to mitochondrial homeostasis via its ability to regulate expression of the mitochondrial kinase Pink-1, whose gene features in Parkinsonism. If replicated, the reports would support the idea that AD and PD molecular pathways share some overlap.
Several LRRK2 mavens contacted by Alzforum expressed skepticism about the conclusion that LRRK2 directly phosphorylates AICD in cells to regulate its activity, in part because the data relies largely on overexpression and inhibitor studies to make the case.
“The investigators used a wide range of techniques, including biochemistry, cell biology, and in vivo work, and the cumulative effect is to suggest there’s something going on here,” Mark Cookson of the National Institutes on Aging, Bethesda, Maryland, told Alzforum. He would need to see additional experiments to prove that APP is a bona fide LRRK substrate in vivo. “People are going to try to replicate this, and hopefully we’ll see other experiments that say yes or no,” Cookson said.
Mutations in leucine-rich repeat kinase 2, a large, multifunctional protein, are the most common genetic cause of Parkinson’s. The best-known mutation, G2019S, boosts LRRK2’s kinase activity, and researchers have tried to pin down which of its many substrates mediate the toxicity of LRRK2G2019S and other mutations (Steger et al., 2016; see also Apr 2014 news; Imai et al., 2008).
To probe whether APP might be such a target, first author Zhong-Can Chen first demonstrated that LRRK2 and APP can interact in cells, by co-immunoprecipitation of either overexpressed or endogenous proteins. When mixed in vitro, LRRK2 catalyzed the phosphorylation of APP at residue 668, a threonine within the AICD. The investigators used a commercially available antibody to phospho-Thr668 APP to detect the modification by western blotting of mouse cortical neuron extracts. The phosphorylation showed up in neurons from wild-type mice, and was enhanced twofold in neurons from the LRRK2G2019S mouse transgenics, a transgenic-overexpressing model. Reducing LRRK2 expression by sh-RNA knockdown in wild-type mouse cortical neurons halved the phospho-Thr668 APP signal, with no effect on APP expression levels.
Phosphorylation of APP at Thr668 has been identified previously in human AD brains and has been said to promote APP processing to Aβ and, possibly, translocation of AICD to the nucleus, where it regulates gene expression (Lee et al., 2003; 2006 comment by Sanjay Pimplikar). To study how LRRK2 might affect the transcriptional activity of AICD, the investigators co-expressed LRRK2 with an AICD-dependent reporter in HEK293T cells. Expression of wild-type or the G2019S mutant both increased reporter gene activity, whereas expression of the D1994A mutant, which lacks kinase activity, or a non-phosphorylatable T668A AICD mutant, did not, suggesting that APP phosphorylation is responsible for the observed transcriptional activation.
To build a case for APP phosphorylation in neurodegeneration, the investigators measured phospho-Thr668 APP levels in LRRK2G2019S mice. At 12 months of age, prior to neuron loss, midbrain phospho-Thr668 APP levels were equal to those in wild-type mice. But by 20 months, phospho-Thr668 APP doubled, accompanied by a decrease in the expression of the dopaminergic neuron marker tyrosine hydroxylase (TH), and slightly fewer TH+ cells in the substantia nigra. With immunostaining, the investigators noted that phospho-Thr668 APP appeared mostly nuclear, suggesting that the antibody picked out AICD that had translocated.
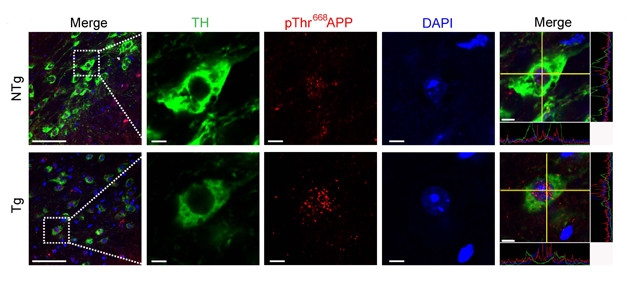
LRRK-ing Danger? APP phosphorylated on Thr668 (red) accumulates in the nucleus of neurons in LRRK2G2019S transgenic mice (Tg, bottom row). [Courtesy of Chen et al., Science Signaling (2017).]
Asking whether AICD might instigate neurotoxicity, the researchers expressed AICD or the non-phosphorylatable T668A mutant in cortical neurons cultured from wild-type or transgenic mice. AICD was toxic to cells from both strains, but more so in the transgenics. In either case, expressing the AICD T668A mutant resulted in less cell death. The researchers repeated the experiment in vivo, by transducing AICD into striatal neurons in wild-type and transgenic mice, with similar results.
Phospho-T668 APP has been detected in human AD brains, and the investigators noted the same in western blots of human PD brain extracts or in dopaminergic neurons derived from iPSCs from patients with LRRK2G2019S mutation.
Finally, they checked how two different LRRK2 kinase inhibitors affected APP phosphorylation. In iPSC–derived DA neurons from LRRK2G2019S mutation carriers, the inhibitor LRRK2-in1 caused a dose-dependent decrease in phospho-T688 APP, and a concomitant increase in TH. In 20-month-old transgenic mice showing dopaminergic neuron loss, treatment with the inhibitor HG-10-102-01 nudged down phospho-T688 APP, and increased TH. However, the short, one-day treatment implies an acute effect on gene expression, not a reversal of neurodegeneration, Cookson noted.
Dario Alessi of the University of Dundee, Scotland, U.K., voiced reservations about the findings. “Based on this data, I’m not persuaded that LRRK2 phosphorylates this AICD domain. If it does, much more work is needed to demonstrate this,” he told Alzforum. “LRRK2 is very sticky; between 300 and 500 proteins have been reported to be co-immunoprecipitated with it. The functional significance of these interactions is questionable,” Alessi said. A key control would be to do the immunoprecipitation with the LRRK2 knockout mouse, show that APP only comes down in the wild-type and not the knockout, and then map the interaction domains on each protein. Alessi would also like to see more characterization of the APP phosphorylation in vitro and in vivo, including in LRRK2 knockouts, with APP T688A mutants, and under conditions of endogenous protein levels.
Cookson said he is concerned that the phosphorylation the authors see in cell lines could be an indirect effect of cell stress caused by overexpression of proteins and activation of stress kinases such as JNK, which is known to phosphorylate APP at T668.
“The thr688 site is a generic target for other interactors, and certainly in an overexpression system the role of JNK and other factors cannot be totally excluded,” Tan responded in an email to Alzforum. “We have demonstrated the LRRK2 phosphorylation of APP in an isolated system in immunoprecipitates, indicating at least that they do interact.”
The finding of neurodegeneration in the older transgenic animals is interesting, Cookson said. This was not seen in the younger, 12-month-old mice, but by waiting longer, the investigators documented a significant 25 percent loss of the dopaminergic neurons. “The field has been struggling to get Parkinsonian features in mice, so to see this neurotoxicity is quite good,” Cookson said.
Whether the phosphorylation is direct or indirect, the results open the possibility of a role for the AICD in PD pathogeneses, said Seth Love, University of Bristol, England, in the U.K. “This is a nice piece of work showing a new mechanism whereby mutants of LRRK2 may contribute to the development of PD,” he told Alzforum. Love praised the authors’ validation of their mouse studies using postmortem human tissues, showing increased APP phosphorylation in LRRK2 mutant carriers. From here, it will be important to understand how AICD causes toxicity in the neurons, and to what extent that is specific for dopaminergic neurons in the LRRK2 mice. Alessi would also like to see some analysis of APP processing, given the documented effect of T688 phosphorylation on BACE cleavage.
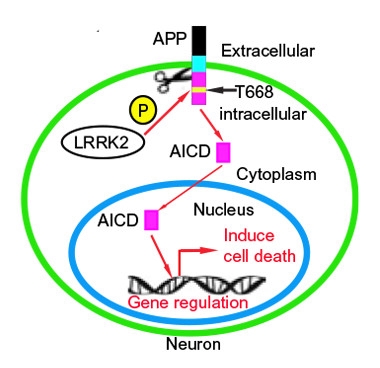
APP in PD.
Schematic of proposed interaction between LRRK2 and APP in dopaminergic cell death. [Courtesy of Chen et al., Science Signaling (2017).]
Bruno Imbimbo, Chiesi Farmaceutici S.p.A., Parma, Italy, wrote in an email to Alzforum, “The AICD has been shown to be involved in a variety of signaling processes, many of which are potentially relevant to AD pathology. … Thus, it would be interesting to know which specific genes are activated upon LRRK2-induced AICD stimulation and if they differ from those induced in AD cellular models.” Imbimbo worked on CHD5074, a small molecule that was being developed until recently for AD and mild cognitive impairment, and that seems to block AICD nuclear translocation and activity. It will be interesting to see whether the compound can protect against LRRK2G2019S-induced neuron loss in PD, the authors write.
In the second paper, lead author Thomas Goiran puts the AICD on a different potential path to neurodegeneration, via regulation of mitochondrial dynamics. AD features mitochondrial deficits, but how these come about is unknown. The investigators noticed that jacking up γ-secretase activity by overexpressing presenilin 1 promoted expression of Pink-1 (PTEN-induced kinase-1). Pink-1 regulates mitochondrial morphology and turnover, and the authors show that AICD controls the expression of the Pink-1 gene in concert with the Foxo3a transcription factor. As a result, either γ -secretase activity or AICD expression influences mitochondrial homeostasis via a pathway that also involves parkin regulation of presenilin 1. The results outline a cascade that involves both Parkinson and AD proteins in the normal physiological control of mitochondria, Alves da Costa wrote in an email to Alzforum. “The parkin-Pink-1 interplay ... mediates a cellular adaptive mechanism by which mitochondrial physiology and dynamics are preserved. This normal control could be altered in both familial and sporadic AD, given that in both cases there is an increase of AICD levels,” she wrote.
“Very little is known about the upstream transcriptional regulation of Pink-1 so the finding that AICD can regulate Pink-1 transcription is significant,” wrote Miratul Muqit, University of Dundee, in an email to Alzforum. “From a clinical perspective, it would be interesting if the authors could show in additional patient derived cells and tissues from AD patients that Pink-1 levels are elevated,” Muqit wrote.—Pat McCaffrey
References
News Citations
Antibody Citations
Research Models Citations
Paper Citations
- Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, Baptista MA, Fiske BK, Fell MJ, Morrow JA, Reith AD, Alessi DR, Mann M. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016 Jan 29;5 PubMed.
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008 Sep 17;27(18):2432-43. PubMed.
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003 Oct 13;163(1):83-95. PubMed.
- Nakaya T, Suzuki T. Role of APP phosphorylation in FE65-dependent gene transactivation mediated by AICD. Genes Cells. 2006 Jun;11(6):633-45. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Chen ZC, Zhang W, Chua LL, Chai C, Li R, Lin L, Cao Z, Angeles DC, Stanton LW, Peng JH, Zhou ZD, Lim KL, Zeng L, Tan EK. Phosphorylation of amyloid precursor protein by mutant LRRK2 promotes AICD activity and neurotoxicity in Parkinson's disease. Sci Signal. 2017 Jul 18;10(488) PubMed.
- Goiran T, Duplan E, Chami M, Bourgeois A, El Manaa W, Rouland L, Dunys J, Lauritzen I, You H, Stambolic V, Biféri MG, Barkats M, Pimplikar SW, Sergeant N, Colin M, Morais VA, Pardossi-Piquard R, Checler F, Alves da Costa C. β-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog-Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol Psychiatry. 2017 May 3; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Chiesi Farmaceutici S.p.A.
Chen and colleagues have proposed a new pathophysiological role for AICD in PD pathogenesis. They showed that LRRK2 phosphorylated APP at Thr668, and that this further promoted the nucleus transcriptional activity of AICD, and its translocation to the nucleus to enhance LRRK2G2019S-induced neurotoxicity.
The AICD has been shown to be involved in a variety of signaling processes, many of which are potentially relevant to AD pathology (Ghosal et al., 2009). The best-known AICD interactor is the adaptor protein Fe65, which stabilizes the AICD and promotes its nuclear translocation (Beckett et al., 2012). In the nucleus, the AICD has been reported to be present within dot-like structures also containing Fe65 and the histone acetyl-transferase Tip60 (Kimberly et al., 2001).
This multiprotein complex is involved in transcriptional activation, though the specific role of the AICD in such a process, its full set of partner proteins, as well as the range of genes it can target are still debated (Hébert et al., 2006). The best-established AICD target gene codes for the pro-apoptotic tetraspannin KAI1, also known as CD82 (von Rotz et al., 2004). Thus, it would be interesting to know which specific genes are activated upon LRRK2-induced AICD stimulation and if they differ from those induced in AD cellular models.
References:
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18367-72. PubMed.
Beckett C, Nalivaeva NN, Belyaev ND, Turner AJ. Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell Signal. 2012 Feb;24(2):402-9. PubMed.
Kimberly WT, Zheng JB, Guénette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001 Oct 26;276(43):40288-92. PubMed.
Hébert SS, Serneels L, Tolia A, Craessaerts K, Derks C, Filippov MA, Müller U, De Strooper B. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006 Jul;7(7):739-45. PubMed.
von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004 Sep 1;117(Pt 19):4435-48. PubMed.
View all comments by Bruno Pietro ImbimboIndiana University School of Medicine
My overall feeling is that this paper is exciting in that it potentially shows a direct interaction between a protein associated with Parkinson's disease, LRRK2, and another protein involved in Alzheimer's disease, APP, thereby suggesting a potentially common pathway between the two neurodegenerative diseases.
This potential linkage between the two diseases is not surprising, because clues for such have been observed for more than 20 years. One of the first clues linking the pathology of Alzheimer's with that of Parkinson's disease was described in by Uéda et al. in 1993, when they sequenced a major protein found in amyloid plaque of Alzheimer's brain samples. They called this protein NACP (for non-amyloid component precursor, and now known as α-synuclein), which a few years later was recognized as the major component of Lewy bodies found in Parkinson's disease brains.
What makes the linkage of LRRK2 with Alzheimer's disease particularly exciting to me is that LRRK2 is an enzyme whose activity we might be able to modulate for therapeutic purposes, thus potentially providing a new avenue for the development of new treatments for Alzheimer's and other neurodegenerative diseases.
A broader implication is that studying other neurodegenerative diseases, including Parkinson's, ALS, and Huntington's disease, also leads to a better understanding of Alzheimer's disease.
References:
Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):11282-6. PubMed.
View all comments by Quyen HoangUniversity of Dundee
Goiran et al. is a potentially interesting study that ties together two major neurodegenerative disorder pathways of Alzheimer's and Parkinson's.
Very little is known about the upstream transcriptional regulation of Pink-1, so the finding that AICD can regulate Pink-1 transcription is significant. That said, the exact mechanism of how AICD controls Pink-1 transcription is unknown. Furthermore, the evidence for effects on mitophagy were lacking as the authors did not employ standard assays to measure mitophagy.
From a clinical perspective, it would also be interesting if the authors could show in additional patient-derived cells and tissues from AD patients that Pink-1 levels are elevated. It would also be exciting to know whether this Pink-1 upregulation is primarily driving AD pathogenesis or is secondary—studying the impact of APP processing in a Pink-1 null background would aid this.
Overall, an intriguing study that will require additional experiments to flesh out the mechanism and clinical significance of these pathways coverging.
View all comments by Miratul MuqitMake a Comment
To make a comment you must login or register.