CONFERENCE COVERAGE SERIES
Clinical Trials on Alzheimer's Disease 2011
San Diego, CA, U.S.A.
03 – 05 November 2011
CONFERENCE COVERAGE SERIES
San Diego, CA, U.S.A.
03 – 05 November 2011
A medical food thought to promote synapse formation was found to improve memory test scores in patients with mild Alzheimer’s disease (AD) in a clinical trial presented at the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD) in San Diego, California, on Friday, 4 November 2011. The 24-week-long Souvenir II trial extends the findings of an earlier proof-of-concept trial, Souvenir I, which lasted 12 weeks (see ARF related news story and Scheltens et al., 2010). Both trials are headed by Philip Scheltens, of the VU University Medical Center in Amsterdam, The Netherlands, who presented the data at CTAD.
The medical food, called Souvenaid, is a product of Nutricia, the medical foods division of Danone Research (which Americans will know as Dannon). It contains a mix of nutrients, including the omega-3 fatty acid docosahexaenoic acid (DHA), uridine monophosphate (UMP), and choline. All are dietary precursors in biochemical pathways responsible for the synthesis of phospholipids, the main components of cell membranes. “We don’t think Alzheimer’s patients are deficient in these nutrients, but we are adding an extra amount to give a boost to these pathways,” Scheltens told ARF. The formulation increased synapse formation in mouse brains and reduced cognitive deficits in transgenic mouse models of AD.
Souvenir II recruited 259 AD patients with a mean age of 74 at 27 European study centers. To be included in the trial, patients had to have a Mini-Mental State Examination (MMSE) score greater than 20, which is considered mild AD. They also could not be taking any medications or nutritional supplements, except for vitamins. The researchers then randomized participants to one of two groups: In the treatment group, 130 people took the 125 ml Souvenaid drink once a day, and in the placebo group, 129 people took another drink with as many calories but without the synapse-promoting ingredients; 118 people in the treatment group and 120 in the placebo arm completed the study.
Patients were examined at baseline, 12 weeks and 24 weeks for various neuropsychological measures. The primary outcome researchers calculated was a composite score of a handful of tests contained in the neuropsychological test battery (NTB). They included the Rey auditory verbal learning test (RAVLT, which measures immediate recall, delayed recall, and recognition performance) and the Wechsler memory scale (WMS) verbal paired associates test (which measures immediate and delayed recall). This primary outcome, which stresses episodic memory, is not one traditionally used in AD studies; Scheltens said it was chosen because more routine test batteries such as the ADAS-cog are insufficiently sensitive to pick up differences in this mild stage of AD.
There was no significant difference in scores between treatment and placebo at 12 weeks. Both improved somewhat, probably due to a learning effect. But by 24 weeks the Souvenaid group had a more favorable trajectory on the memory score than controls, leading to a Cohen's d effect size of 0.22 points that Scheltens said was significant. There were, however, no differences between drug and placebo on the trial’s secondary outcomes resulting from the total NTB, such as executive function domain, total composite score, and individual item scores. Souvenaid had no significant side effects, Scheltens reported.
Despite these favorable findings, it is too early to know what they mean for AD patients. “There is not enough evidence at this point to make a recommendation to patients,” said Paul Aisen at the University of California, San Diego. Aisen heads the Alzheimer's Disease Cooperative Study, which hosted the conference.
Although Souvenaid showed some benefit in mild AD, it failed to do so in more advanced disease. A parallel clinical trial to Souvenir II recruited 527 people with mild to moderate AD (MMSE scores 14-24) at 48 U.S. clinical centers. Unlike patients in the Souvenir II trial, who were not receiving any medications, these U.S. patients were taking cholinesterase inhibitors and/or memantine. At 24 months, there was no difference between the treatment and placebo group in 11-ADAS-cog scores, which was the primary outcome measure in this trial. “We think this supplement works by building membranes and synapses, but it only works if there are enough neurons there. Patients with more advanced AD may not have a sufficient number of neurons, and that is why we saw no effect,” Scheltens speculated.
Scheltens said the results warrant further studies, and two are ongoing. The group is conducting another trial in moderate AD patients and one in prodromal AD as defined by the “Dubois criteria” (see ARF related news story on Dubois et al., 2007). The prodromal AD study, funded by the European Union, will enroll 300 patients and last 24 months, with some centers extending to 36 months.
Medical foods are prescribed to patients by their doctors, but they are not typically tested as drugs since they do not have to go through the same regulatory process (see ARF news series on medical foods in AD). They have to show safety but not efficacy. Even so, the sponsor decided to mount a clinical trials program in an effort to build support among physicians. “If doctors are going to prescribe Souvenaid, I want them to have all the scientific information,” said Scheltens.
Other clinicians at CTAD were encouraged that there was a program of studies conducted on Souvenaid. They noted that the present trial in drug-naïve mild AD patients seemed as rigorously designed as trials of pharmaceuticals and cleanly met its prespecified primary endpoint. They also cautioned that the primary endpoint indicates an improvement at six months only in episodic memory, not in the much broader swath of symptoms that define AD. Others recalled that previous trials of individual components of this nutrient mixture have been negative (see, e.g., ARF related news story on DHA trial).
Lon Schneider of the University of Southern California, Los Angeles, noted that, because the bulk of preclinical research on the drink’s ingredients was done on cognitive aging rather than AD, a trial in age-related cognitive memory impairment might be informative. Schneider added that a robustness analysis looking at whether the participating centers contributed about equally to the effect on the NTB would be of interest. Other trialists said their confidence in the effect would grow once other evidence supporting Souvenaid’s effect on human synapses became available, such as FDG-PET, functional MRI, or encephalography (EEG).
In general, synaptic activity is getting more attention in AD clinical studies. At CTAD, a number of studies examined the use of event-related potentials as measured by EEG. Such measures of synaptic function could serve as markers of early AD as well as to measure disease progression and even treatment response. “More studies are looking at functional connectivity using functional MRI and EEG,” said Scheltens. “There is an awareness that early in disease, the network gets disrupted, so improving connections is a way for treating the disease.”
Scheltens and colleagues have collected EEG and magnetoencephalogram (MEG) data from a subset of Souvenir II participants; their analysis is underway. Scheltens did not present these results at CTAD, but once available they may provide further understanding on how Souvenaid acts.—Laura Bonetta and Gabrielle Strobel.
Another drug bites the dust. On the opening day of the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD), held 3-5 November 2011 in San Diego, California, Douglas Galasko, of the University of California, San Diego, presented data from a randomized clinical trial of PF-04494700, a small-molecule inhibitor of RAGE (aka receptor for advanced glycation endproducts). The trial was halted before its intended conclusion, as interim data did not show there would be a benefit. Together with the National Institute on Aging, the drug’s sponsor, Pfizer Inc., co-funded this Phase 2b trial through a contract with the Alzheimer's Disease Cooperative Study (ADCS), which hosted CTAD this year. Pfizer has discontinued further development of this drug, which the company had previously tested for type 2 diabetes as well.
Galasko’s presentation offered a glimmer of hope when he described results of a follow-up examination, conducted after treatment was suspended. That analysis hinted at a possible belated clinical benefit for a low dose of the study drug; however, that only became evident after most patients had already been taken off the treatment. Although far from conclusive, the findings may serve as a cautionary tale for adaptive-style trials that rely on interim analyses, said Galasko.
RAGE is a cell-surface receptor of the immunoglobulin superfamily. It binds advanced glycation endproducts (AGEs); these are modified forms of lipids and proteins that become glycated when exposed to sugars. When bound to their receptor, AGEs, which form during normal aging and in higher amounts in patients with diabetes, cause inflammation and oxidative damage to cells. Recent work has shown that RAGE also binds amyloid-β (Aβ) and mediates toxic effects of Aβ oligomers in neurons (see ARF related news story) The PF-04494700 RAGE inhibitor blocks this interaction, so the hope was that it would provide a combined treatment effect across inflammatory and amyloid-related processes. In preclinical studies, the compound decreased brain Aβ load in transgenic mice and improved their performance on behavioral assays. A prior 10-week-long Phase 2a safety trial showed a good safety profile of this drug in patients with AD, but no changes on clinical measures (Sabbagh et al., 2011).
The trial Galasko described at CTAD (see on ClinicalTrials.gov) was a proof-of-concept trial designed to test safety, tolerability, and efficacy of two doses of PF-04494700 compared to placebo over 18 months. It enrolled 399 AD patients randomized to one of three groups: The first (135 patients) received 20 mg per day of the drug, the second (132 patients) received a 5 mg dose, and the third (132 patients) were on placebo. All patients were over age 50, with Mini-Mental State Examination (MMSE) scores between 14 and 26. Many patients were on AD medications, such as acetylcholinesterase inhibitors or memantine, but not on diabetic or immunosuppressive drugs, as these might interfere with RAGE’s mechanism of action.
The researchers calculated ADAS-cog (Alzheimer's Disease Assessment Scale-cognitive subscale) scores as the primary outcome measure, along with various safety indications. As secondary outcomes, they conducted additional cognitive tests and collected data on structural magnetic resonance imaging (MRI) measurements, Aβ imaging using positron emission tomography (PET), and levels of cerebrospinal fluid (CSF) biomarkers Aβ and tau on subsets of patients.
According to the trial’s design, researchers were to take measurements at baseline and every three months thereafter up to 18 months. The trial was not Bayesian, but it did include some adaptive features—mainly, it could change course based on the results of two interim analyses conducted by the Data and Safety Monitoring Board (DSMB), an independent group of experts that advises trial investigators.
The first interim analysis, which was conducted six months after half of the intended subjects had been randomized, was purely a safety analysis. It showed that patients receiving the higher dose of the drug had more serious side effects, and their ADAS-cog scores worsened more quickly compared to the other two groups. As a result, that arm of the trial was stopped. The low dose was safe. The second interim analysis, conducted a year later, was a futility analysis to assess whether the low-dose treatment appeared to be providing a prespecified level of benefit. It narrowly missed that level; hence, all patients were taken off the medication and the trial was essentially over. But researchers continued to follow participants for one more visit, up to three months later, to learn more; about half of the original patients completed a last visit at 18 months.
When Galasko and colleagues analyzed the complete dataset including all 18-month measurements, things got interesting (or confusing, depending on one’s point of view). They found that the rate of cognitive decline in the high-dose group had slowed down, and their ADAS-cog scores were now similar to those of the placebo group, at least for those patients who continued to return for follow-up visits. Galasko could not explain the faster cognitive worsening in this group or their subsequent stabilization of cognitive symptoms. “We don’t know the mechanism of toxicity. We did not find any evidence of vasogenic edema or any other abnormality on MRI,” he said.
When the researchers analyzed the results of the 69 patients in the lower-dose group who completed the 18-month analysis, they found post-hoc evidence of improvement in their ADAS-cog scores compared to the 68 patients in the placebo group. There were, however, no differences in other clinical outcome measures or in the rates of hippocampal shrinking or CSF biomarker measures. This improvement in ADAS-cog scores was not detected at the 12-month timepoint, and it disappeared if the researchers only analyzed test scores obtained from patients while they were receiving the study drug.
Although the clinical trial did not provide evidence to support continued development of PF-04494700, Galasko said ending the trial early may have prevented the scientists from seeing any positive effects that required more than 12 months to become apparent. “We need to be careful about adaptive designs and stopping rules if we want to get the maximum amount of information about a drug,” he said. Other researchers at the meeting pointed out that the findings are difficult to interpret, given that the trial was stopped early and so many patients dropped out. This is the second investigational AD treatment Pfizer discontinued recently (see ARF related news story).—Laura Bonetta.
Paul Aisen heads the Alzheimer's Disease Cooperative Study (ADCS), a federally funded clinical trials consortium headquartered at the University of California, San Diego. The ADCS hosted the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD) from 3-5 November 2011. In his opening lecture, Aisen narrated the history of the field’s attempts to develop treatments for Alzheimer’s. How did it start, how did it get stuck in its current barren phase, and how can it regain traction? Alzforum asked Aisen to give his lecture a second time on our Webinar platform so that the field at large could hear it. If you are a student, or a basic scientist who is becoming interested in translation, or a newcomer to the field, then this lecture will bring you up to speed on what has been done so far and where the cutting edge has moved. If you are a veteran trialist, you might enjoy an opportunity to revisit milestones of days past, chew on challenges of days present, and look to a future of secondary and, ultimately, primary prevention of AD.
Did you know, for example, that the first clinical AD trial, by William Summers (Summers et al., 1986), was of such questionable quality that it embroiled the New England Journal of Medicine in controversy and led to a fraud investigation by the Food and Drug Administration and the respective academic medical center? Or that this initial trial was followed by two negative trials, at which point attempts at cholinergic treatment might have ended? For that matter, few of the earliest trials would pass methodological muster today. Despite those shortcomings, however, the early trials did focus attention on the goal of trying to develop drugs for what was considered an untreatable disease. They sparked larger, controlled, multicenter trials that led to the symptomatic drugs that are the standard of care today.
Beyond recalling historical vignettes, this lecture also offers a serious account of a current dilemma. Aisen discusses why the previously workable Phase 2 model for symptomatic drugs proved ill suited to give reliable efficacy signals for disease-modifying drugs that do not detectably improve symptoms in the short term. He reviews the failure of some of the large, long, expensive Phase 3 trials for nine different drugs in the past decade.
Clearly, better drugs are needed, but the failures have more than one reason. For their part, what can trialists do? Aisen says they can sharpen their tools to test drugs earlier, and in this way, get ready to act on building international consensus that AD starts some 15 years before symptoms. Previous trials in mild cognitive impairment (MCI) have failed, and Aisen dissects the reasons why that might be. He moves on to new trial designs based on a biomarker-supported definition of prodromal AD, which are garnering nods from the FDA and its European counterpart, the European Medicines Agency (EMA). Prodromal AD, unlike MCI, is considered a treatable disorder, Aisen says, and that opens up trial designs of greater statistical power. Several companies have begun using such designs.
But what if prodromal AD proves still to be too late? In the second part of the talk, Aisen speaks about laying the groundwork for secondary prevention trials. Those would be for people who are coming to clinics spurred not by memory problems, but for cognitively normal older people who have been studied and show the signature of pathological biomarkers that is thought to define preclinical AD. Regulators have been encouraging, saying, for example, that such trials could use a combination of a cognitive endpoint plus a panel of biomarkers as outcome measures, doing away with the requirement for a global clinical outcome.
Aisen wraps up with remarks on primary prevention, that is, trials in people in midlife who are normal not only cognitively but also in their biomarker profiles. Primary prevention aimed at staving off a pathological change of those early markers is out of reach at this point in time. Even so, Aisen calls on the field to think ahead and develop the biomarker and aging research necessary to make it feasible.
Play the Webinar. Questions? Comments? Add your perspective in the comment box below.—Gabrielle Strobel.
No Available Comments
Here is a typical scenario. A big trial ends, the sponsor announces success or failure, the researchers describe the findings at conferences, and if the press takes an interest in the story, the results may end up on the evening news. But throughout the process, we seldom hear from the patients who participated in the study and their caregivers, or the hospital staff who administered the tests. What did they think about the trial? Two presentations at the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD), held 3-5 November in San Diego, California, asked precisely that question. The answer, put simply, is that Alzheimer’s disease (AD) trials are extremely burdensome, sometimes unnecessarily so, and hard to comply with to the bitter end for most elderly people. Another CTAD presentation suggested one solution: home-based assessment methods that relieve some of the burden.
“In the earlier days of AD studies it was very straightforward: three standard memory scales, one to two activities of daily living scales, blood samples and electrocardiograms for safety, and one CT scan at study entry. Subjects visited every three months for half a day or less and everyone was happy,” wrote Roseanne Hogarth, a research coordinator at Hornsby Ku-ring-gai Hospital in Australia, in an e-mail to ARF. At CTAD, Hogarth presented a poster bluntly titled “Protocol from Hell.” It described the frustration of volunteers, caregivers, and research coordinators with the growing complexity of AD trials. Today, AD patients are asked to fill out several lengthy questionnaires testing their cognition; those paper-and-pencil sessions are increasingly accompanied by magnetic resonance imaging and/or lumbar spine punctures to collect cerebrospinal fluid. In addition, researchers usually collect blood samples, and may also take electroencephalography or other measurements. “The designers of these protocols are making full use of the advances in technology, but forget there is a person with dementia in the study who may have very limited abilities,” wrote Hogarth. Study visits for such protocols can take up to eight hours every four weeks, Hogarth added.
In a survey of some 17 principal investigators and study coordinators and 25 trial participants and caregivers at different sites in Australia, Hogarth and Susan Kurrle collected opinions about current AD study procedures. The main complaints from people conducting trials were that informed consent forms are far too long and contain complex language and concepts that are too difficult for people with dementia to understand. Trial staff bemoaned having to undergo continual retraining on tests with which they are familiar, mainly because each sponsor made slight changes to the Standard Assessment Scale. They warned that participants have to do so many procedures per visit that they leave exhausted. In addition, trial staff said, some of the psychometric tests used in protocols are unsuitable for people with significant cognitive impairment. When caregivers and patients were given a chance to weigh in, they mentioned that trials involve too many study visits, cutting into holidays or other activities. They found visits too long, requiring reorganization of weekly routines, and complained that trials involve repetitive questionnaires with redundant information. Study participants also complained that instructions about study drugs are often too difficult to understand.
Besides reducing efficiency and bumping up cost, demanding protocols make it harder to recruit and retain participants. “Some studies are so burdensome, people refuse to join,” wrote Hogarth.
To look more closely at what determines whether someone says yes or no to a trial, Sandrine Andrieu, Bruno Vellas, and colleagues at the University of Toulouse in France conducted...well, a trial. The ACCEPT study followed up, with questionnaires and in-person interviews, both people who accepted and other people who refused participation in an intervention study for the prevention of AD, the Multidomain Alzheimer Preventive Trial (MAPT; see Gillette-Guyonnet et al., 2009). The impetus for the ACCEPT trial, explained Andrieu in her presentation at CTAD, was the notion that people who participate in clinical trials for AD are not necessarily representative of the general population. That discrepancy might help explain why some protective factors identified in epidemiological studies fail translation into interventions in clinical trials. For example, Andrieu mentioned that in 1999, about 12.2 percent of people 65 years and older in France had a high school diploma, yet among participants in the Three-City Study recruited around that time, that number is 22 percent. In 2010, the level of high school-educated French elderly people rose to 17.6 percent, but was 45.4 percent among MAPT participants. Such skewing might affect results, as higher education is a protective factor for AD (see ARF related news story).
In Andrieu’s study presented at CTAD, out of 1,902 cognitively normal people 70 years and older who agreed to participate in the MAPT, 71 percent also completed the ACCEPT questionnaire; 54 percent of the 674 people who declined to join MAPT did the ACCEPT questionnaire. Twenty people from each group were followed up in semi-structured interviews. From those and the questionnaires, Andrieu and colleagues identified several factors that distinguished MAPT participants from the "thanks, but no thanks" group. The older people were, the more likely they were to refuse participation. People who had never married were less likely to participate. People with higher education and high incomes were more likely to participate. People who thought they might be at higher risk of AD, for example, those with a family history of the disease, were more likely to participate.
It made no difference to participation whether people heard about a trial from a relative or friend or the media, or whether someone had a big or small social network. However, one key factor in their motivation was the nature of the relationships patients had with their doctors. The more confidence people had in their doctors, the more likely they were to participate in the prevention trial, Andrieu reported.
The main reason people gave as wanting to participate in the trial was that they were interested in research and wanted to help those with the disease. Some people who participated thought the trial would help them find ways of training their memory and thus benefit them. On the other hand, people who refused said the trial was too long or did not fit into their schedules. Some people had problems with transportation or did not want to go to a hospital. Others refused because they could not choose whether they would end up in the placebo or treatment arm, Andrieu reported.
Perhaps it should not be surprising that people are loathe to go to the hospital, especially for prevention trials whose participants are not sick. Then one way to make clinical trials more palatable to a wider group might be to carry out assessments and tests in their homes. That notion is being tested in a large trial sponsored by the Alzheimer's Disease Cooperative Study (ADCS). The Home-Based Assessment (HBA) Study, which completed enrollment in 2009, recruited 640 cognitive normal participants with a mean age of 80 and older who lived at home. They are required to take a multivitamin twice a day for four years while completing cognitive assessments at monthly, quarterly, or yearly intervals. These work by way of one of three home-based methods. One group of participants mails in questionnaires and answers questions in telephone interviews with a real person; they keep track of their vitamin intake in a written log that they also return by mail. The second group conducts assessments via an automated phone response system, and vitamin compliance is monitored with telephone keypad responses. The third group is the most high tech. Here, participants use desktop computers equipped with touch screens and speech recognition software to record answers. They also use the Med-Tracker, an electronic pillbox that automatically records when they remove a vitamin. Mary Sano of Mount Sinai School of Medicine in New York City, who co-directs HBA together with Jeffrey Kaye of Oregon Health and Science University in Portland and Steven Ferris at New York University in New York City, told ARF the trial was able to recruit 20 percent minority patients. “This study was designed to try to recruit a sample that is more representative of the population at risk for AD,” said Sano. “We hope the results will develop interest in more studies being home-based.”
“Right now, to participate in a study, you have to be able to travel to a clinic. That’s only possible for people who live near a large academic hospital or university, and those who can drive or have a caregiver who can drive,” said Kaye.
That is not to say that home-based assessments are without complications. Some elderly people live in apartments too small to accommodate a computer; others live in areas that have no broadband or Internet access. In some cases, Kaye said study coordinators did not know how to set up the system.
Sano and Kaye are trying to reduce the burden on patients on all fronts. “We assess several domains of cognitive function, and we try to cut down the number of items in each assessment to eight per domain,” Sano said. “This is huge, because right now tests are too cumbersome.” The scientists are also trying to determine just how often to assess participants. “Doing assessments more frequently gives you more intimate data, but may lead to more dropouts. Patients don’t like doing the assessments too often. But if you do them too infrequently, the danger is that participants can lose interest in the study,” Sano explained.
Beyond getting people to join and stay with the study, home-based monitoring may also provide richer and more accurate data, Kaye argued in his presentation at CTAD. When answers are recorded by a phone or computer system, they are automatically analyzed. “There is less opportunity for data artifacts that arise from different people scoring the answers,” he said. Audio files can be further analyzed for pauses between answers or other speech features that may provide insight into cognitive function. Kaye’s group has also developed door sensors that can monitor how often people go out or open the fridge, and infrared sensors installed on ceilings that can measure a person’s walking speed during everyday activities. Some studies have shown that changes in how quickly a person walks can serve as early markers for mild cognitive impairment (MCI) (Hayes et al., 2008). “Traditional measures typically capture only a snapshot of cognition in an artificial setting,” said Kaye. “Home-based systems allow for more continuous real-time assessments.”—Laura Bonetta.
No Available Comments
Regular visitors to Alzforum will be familiar with the use of positron emission tomography, magnetic resonance imaging, and cerebrospinal fluid measurements as markers for Alzheimer’s disease (AD). But these three are not the only AD biomarkers. There is also the humble event-related potential (aka ERP). Determined by electroencephalography (EEG), this lesser-known electrophysiological measurement has been a relative wallflower, but is now gaining some popularity among AD trialists. At the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD) held 3-5 November 2011 in San Diego, California, ERPs had a symposium and some posters devoted to them. “As a tool for assessing cognitive function, ERPs have been around for a long time, but mostly in the realm of scientists doing basic studies of memory and other processes rather than in the realm of trialists,” Andrew Budson of Boston University, told ARF. Budson jointly organized the ERP symposium at CTAD with Lon Schneider of the University of Southern California, Los Angeles.
ERPs have remained below the radar in clinical studies, Schneider told Alzforum. “AD researchers have been relying on biomarkers like CSF proteins and brain imaging. They have not had the motivation to use EEG,” he said. The lack of new therapies for AD and drug failures in late-stage clinical trials may now be providing that motivation, for example, to try to develop a cheaper, more immediate neurophysiological indicator of drug effect, he added.
What are ERPs, anyway? They represent EEG recordings elicited in response to particular stimuli. One common example is the “oddball” paradigm, in which researchers ask people to distinguish an infrequent, “oddball” tone (say, a high-pitched 2,000 Hz sound) among more frequent 1,000 Hz tones. This paradigm generates EEGs that, when averaged to cancel out random variation, form a reproducible wave pattern of neuronal activity, or the ERP. Its components—each being a discrete peak or trough of few microvolts in magnitude—represent summed inhibitory or excitatory postsynaptic potentials. They are designated as N or P, depending on whether they are negative or positive, followed by a number that indicates their timing (or latency).
Each ERP component results from one or more mental operations occurring at a specific time after the stimulus, with a set amplitude and distribution across a person’s scalp. Some components have standing names and are well known to scientists. For example, the auditory oddball paradigm elicits early components, such as the N100 (or N1) and P200 (P2), which represent activity in the first cortical areas to receive and evaluate the sensory input. Later components of the same ERP, such as the intensely studied P300 (P3) component, represent the processing of information at more advanced cognitive levels. Scientists think that the P300 elicited by oddball stimuli reflects the working memory updating mental representations. Still later components of the ERP (i.e., N400 or P600) involve cognitive functions that have to do with the processing of language and syntax, as well as memory.
There is a substantial literature of some 370 papers showing that various ERP components are altered in either amplitude or latency in AD patients, and that ERPs could thus be used as diagnostic markers of the disease. The first paper from Floyd Bloom’s group at The Scripps Research Institute in La Jolla, California, showed that the P300 component elicited by an oddball auditory stimulus was smaller in amplitude and had a longer peak latency in AD patients than in normal controls (Polich et al., 1990).
But the value of ERPs in AD research was only recently appreciated with the recognition that AD is a primarily synaptic disorder. In 2002, Dennis Selkoe at Brigham and Women’s Hospital in Boston, Massachusetts, drew attention to prior findings by others on synapse loss early in AD, and proposed that amyloid-β oligomers cause synaptic dysfunction before the deposition of amyloid plaques in the brain (Selkoe, 2002). Nine years later, researchers updated the model of biomarker changes associated with AD development (see ARF related Webinar) to include changes in synaptic function as one of the earliest markers in the preclinical stage of AD, occurring before tau-mediated neuronal injury or brain structure changes (Sperling et al., 2011).
Because ERPs provide a sensitive measure of synaptic function, they can provide clues into the early steps of AD, argued John Olichney, University of California, Davis, in his presentation at CTAD. ERPs constitute a millisecond-by-millisecond record of neural information processing; this stands in contrast to a temporal resolution of seconds to tens of seconds with functional MRI or PET. On the other hand, “ERPs are an older technique with limited spatial resolution compared to current neuroimaging methods,” wrote Olichney in an e-mail to Alzforum. That might be one reason they have been underutilized in clinical studies. Another is that “clinical validation of these sensitive measures has been slow and difficult work,” Olichney added.
ERPs as Diagnostic and Prognostic Markers
Two talks at the CTAD symposium showcased the clinical value of ERPs in predicting risk of AD development. The first, by Olichney, focused on changes in the N400 and P600 responses. Olichney’s group has previously developed a common paradigm for testing them. It begins with giving patients several phrases, each describing a category, for example, “a breakfast food” or “a continent.” Subsequently the patient hears a word that is either congruous (i.e., “waffle” following the breakfast food phrase) or incongruous (i.e., “table” following continent) with its preceding category. In healthy people, new congruous words elicit large P600 and N400 amplitudes, compared to incongruous words. Once the congruous words are repeated, i.e., no longer new, the N400 and P600 amplitudes shrink relative to their initial values; this shrinking is referred to as the “repetition effect.” Olichney and others have reported that the repetition effect is less apparent in mild AD patients than in healthy people (Olichney et al., 2006).
In a study on 32 patients with mild cognitive impairment (MCI), Olichney showed that abnormal ERP word repetition effects provide a marker for high risk of progressing to AD. MCI patients who had abnormal repetition effects (either N400 or P600) at baseline had an 87 to 88 percent likelihood of developing dementia three years later (Olichney et al., 2008). “The hazard ratio of the risk prognosis compares favorably with MRI and FDG-PET in predicting AD progression from MCI as established by ADNI [the Alzheimer’s Disease Neuroimaging Initiative],” said Olichney at CTAD (see ARF related news story).
For his part, Karim Bennys at Montpellier University Hospital in France described comparable data focusing on the P300 ERP component. This component can be broken into P3a, which measures focal attention originating from the prefrontal cortex, and P3b, which measures working memory. Bennys examined 71 patients of mean age of 71 years diagnosed as having MCI on the basis of the Petersen criteria (Petersen, 2001), compared to 31 cognitively normal controls with similar age and education level. These people were examined at baseline and again one year later using an auditory oddball paradigm.
One year later, 41 people in the MCI group were defined as progressors because their executive function had declined significantly compared to the 30 non-progressors. At baseline, the progressors showed increased N200 and P3b latency and lower N200 and P3b amplitudes than non-progressors. In the progressors, the gradient with which the P3b appears from the front to the back of the head was inverted, with a significant decrease of amplitude in the parietal cortex compared to non-progressors. By this point in time, 17 of the progressors met clinical criteria for AD. “The modification of N200 and P3b amplitude and latency appear very early in MCI patients at high risk of progressing to AD,” Bennys said at CTAD. He added that P3b amplitude alone is a robust marker for the conversion from MCI to AD, with 80 to 90 percent sensitivity and 70 to 80 percent specificity.
As the AD field is moving toward conducting trials in mildly or even presymptomatic people (see ARF related news story and ARF related Webinar), a recent paper has shown the value of ERPs in this group. A study by the Alzheimer's Prevention Initiative (see ARF conference series) used high-density ERPs to examine brain physiology in 10 presymptomatic carriers (average age of 34 years) of the E280A mutation in the presenilin-1 gene (Quiroz et al., 2011). These people performed a visual recognition memory test during which ERPs were recorded, and the recordings were then compared to those taken from 11 siblings without the mutation. The results, published this year in Neurology by first author Yakeel Quiroz at Boston University and the University of Antioquia in Medellin, Colombia, showed that despite identical test performance, mutation carriers had smaller amplitude from 200 to 400 ms in the frontal brain regions and a higher amplitude in the occipital regions than did controls (Quiroz et al., 2011). “I hear from our Colombian colleagues that one advantage of ERP biomarkers is that not everyone can afford to do a PET scan, but everyone has an EEG recorder,” said Budson, who is senior author on the paper.
Budson, Olichney, and others are in the process of looking at MCI and AD patients to determine which paradigms and ERP components provide the best measures for clinical diagnosis. “It may be that we will develop diagnostic markers made up of one or two different paradigms, or that some paradigms will be more or less sensitive for different stages of disease,” Budson told ARF.—Laura Bonetta.
This is Part 1 or a two-part series. See also Part 2.
No Available Comments
At the 4th International Conference on Clinical Trials on Alzheimer's Disease (CTAD) held 3-5 November 2011 in San Diego, California, attendees got an earful about an arguably neglected topic in Alzheimer’s research, that is, the use of event-related potentials (ERP) as biomarkers of disease. Part 1 of this series covered how these measures of cognitive function taken by electroencephalography (EEG) were reported to have predictive value in staging AD. But CTAD also featured talks on another application of ERPs that appeals to scientists. It is their use in determining whether a drug is hitting the desired target. “Instead of testing a drug in 100 people to see if after 18 months there are any clinical effects, you can treat a smaller number of patients and look for small differences in ERPs that would indicate target engagement,” said Lon Schneider of the University of Southern California, Los Angeles. EEG potentials have been used in drug development for anti-epileptic and psychiatric drugs for many years to see whether a drug is affecting central nervous system function, but they have not been widely used in AD. Part of the motivation for revisiting ERPs in AD drug discovery comes from recent failures in AD clinical trials. “People are going back and trying to re-evaluate the tools they are using,” Schneider told ARF.
For ERPs to help drug development, they need to be characterized in animal models of disease, where drugs are initially tested. In his talk at CTAD, Steven Leiser, Lundbeck Research, Paramus, New Jersey, described his work in determining whether there is a rodent equivalent of P300. “Are rats that smart?” he asked the audience. Backtranslating a common ERP paradigm from human P300 research into rodents, Leiser developed a test in which rats had to discriminate a target tone from more frequent distracter tones; they then had five seconds to poke a niche with their noses. If they got it right, they received a food reward. The rats learned to perform this task by day six. Leiser and colleagues took EEG recordings during the task and then studied the ERPs elicited by the target tone. By day six, the researchers could clearly detect an ERP component in the rats’ recordings whose timing correlated with that of the human P300. The amplitude of the rat P300 correlated with how well they performed the tone recognition task, Leiser reported.
In addition, the rat P300 recapitulated some of the pharmacological attributes of the human counterpart. When the experimental anticholinergic agent scopolamine is given to healthy individuals, it impairs their memory and reduces their P300 response in a way that mimics, for experimental purposes, some effects of AD. Donepezil, an acetylcholinesterase inhibitor drug that many AD patients take, counters scopolamine’s actions. Sure enough, when Leiser gave scopolamine to the rats, the agent disrupted the rat P300, and subsequent donepezil administration rescued it.
Changing direction back to the usual trajectory of translational research—that is, from rats to humans—Niclas Brynne of AstraZeneca provided an example of how his group deploys ERPs to gauge a drug’s efficacy in a human drug trial. He presented results from a Phase 2 multicenter, randomized, double-blind, placebo-controlled crossover study to evaluate the pharmacodynamic effects of single and multiple oral doses of an agonist of the α4β2 subtype of the neuronal nicotinic acetylcholine receptor. The trial compared the effects of the drug, called AZD1446, versus placebo and a single dose of donepezil on quantified EEG (qEEG)—these are measurements of spontaneous brain waves—and ERPs in 40 patients with early AD (MMSE scores of 18 to 24) over the course of nine weeks. (For a status update on this drug, see ClinicalTrials.gov.)
In AD patients, the power of α-type brain waves detected by qEEG is reduced compared to that in cognitively normal, healthy people (see ARF related news story). At CTAD, Brynne presented data suggesting that both AZD1446 and donepezil ameliorate these changes in α rhythms and in a slow wave index seen in AD patients. The drugs also reversed the patients’ defects in P300 latency and amplitude. These findings “support target engagement by AZD1446 with possible positive effects on cognition,” Brynne said at CTAD.
Although ERPs are not in the mainstream of AD drug development, their moment on center stage at CTAD may signal a change in the field. “We have shown in the symposium that ERPs can provide value, particularly when we are in a situation in which new drugs are not proving successful,” Schneider told ARF. “Many drugs are failing in Phase 2 and 3, so we are asking ‘Could we have predicted those failures earlier using different markers?’”—Laura Bonetta.
This concludes a two-part series. See also Part 1.
No Available Comments
In the second half of 2011, scientists driving the Alzheimer's Prevention Initiative have been reporting at scientific conferences the first emerging biomarker findings from their human volunteers. These data provide tantalizing glimpses of what happens in the brains of young people carrying a deterministic Alzheimer's disease mutation when they are still in their twenties and thirties. While these imaging and fluid data at present represent but small snapshots of the disease 25 years before dementia, they nonetheless suggest that a quiet drama unfolds in the Alzheimer's-bound brain years before amyloid. “At present, it looks as if functional and structural changes may occur prior to fibrillar amyloid deposition,” Adam Fleisher of the Banner Alzheimer’s Institute said in a talk at the Clinical Trials in Alzheimer’s Disease (CTAD) conference held 3-5 November 2011 in San Diego, California. If further data substantiate those initial findings, and if the findings generalize to late-onset Alzheimer’s, they would then call for a refinement of the proposed biomarker staging diagrams that have captured the imagination of Alzheimer’s disease researchers worldwide.
Fleisher belongs to a large collaborative team of scientists who have been developing the API as a program meant to pioneer secondary prevention trials in people who are at high risk of developing Alzheimer’s disease. Led jointly by Eric Reiman and Pierre Tariot at the Banner Alzheimer’s Institute in Phoenix, Arizona, and Francisco Lopera at the Universidad de Antioquia, Medellin, Colombia, the API has been doing the groundwork preparing for such trials in people who carry autosomal-dominant mutations that will give them the disease with near certainty. (The API also prepares for trials in aging people who carry the ApoE4 risk allele.) “There are many people who are at very high risk of AD who are clamoring for therapeutic trials,” Tariot said.
The Initiative’s autosomal-dominant half is complementary to the Dominantly Inherited Alzheimer Network (DIAN, ARF related story), and its late-onset half is complementary to the A4 initiative. Together, the three programs share the goal of breaking ground on secondary prevention drug trials across the AD spectrum. That is, they range from rare, deterministic AD genetics on one end to risk genetics in the middle, and to the most common forms of late-onset AD on the other end. Success in any and all of these trials could energize earlier-stage trials throughout the field, the scientists believe. However, each program is also unique in some aspects. DIAN has fewer patients than the API, but subsumes all APP and presenilin mutations; A4 is potentially the largest study, but further behind in terms of funding and driven by biomarkers, not genetics. Along the way of gathering observational data and planning their respective programs, the leaders of all three meet frequently to work out where they can coordinate to enhance each other’s goals and ensure that their respective datasets can be analyzed together.
So what’s new with API since its last update on Alzforum (see ARF API series)? In 2011, the researchers have enrolled some 1,300 relatives of the Colombian families afflicted with the E280A Paisa mutation in presenilin 1 into the observational biomarker and cognitive study phase meant to precede treatment trials. About a third are carriers. The scientists hope to bring the number of participants to 3,000 and the number of carriers close to a thousand by 2013.
That goal—as indeed all key goals of API, DIAN, and A4—hinges on new funding coming forward. In the case of API, its leaders are currently awaiting final review by the National Institute on Aging of a pending grant proposal for the first treatment trial with an identified (but undisclosed) experimental drug while simultaneously stitching together a funding coalition of company money and private philanthropy.
In the meantime, the scientists have expanded their original biomarker studies with the Colombian participants that started in 2010. In 2011, the scientists, led by Fleisher and Yakeel Quiroz, currently at Boston University, added new cohorts of cognitively older adults in age brackets from age 35 and up, all the way back to children aged eight to 17. The children are not undergoing spinal taps, but they are donating a blood sample and, importantly, lying still in the scanner for various modalities of magnetic resonance imaging.
Why children? The scientists want to chronicle the entire natural history of this form of AD from its beginning, meaning they will trace back at what age biomarker measurements begin to diverge between carriers and their non-carrying siblings. In the next-older age bracket—the 18- to 26-year-olds—mutation carriers already show distinct differences in brain function and even structure. Hence, Quiroz and colleagues reached back with the less invasive tests into even younger ages.
To date, MRI has been taken from some 200 volunteers age eight and up. This happens on a Siemens 1.5T scanner at the Hospital Pablo Tobón Uribe in Colombia. “MRI capability is very good there for API studies,” Tariot told the audience at CTAD. Plasma has been taken from some 130 volunteers age eight and up, CSF from some 90 people age 18 and older. Fluids are being drawn in Medellin following standard acquisition, preparation, storage, and shipping directions developed for DIAN. They are analyzed in the lab of Anne Fagan at Washington University, St. Louis, Missouri, to ensure that data are comparable with CSF measures in the DIAN and, indeed, the Alzheimer’s Disease Neuroimaging Study (ADNI). PET imaging with florbetapir started up in September, when the first of what will be 50 participants flew to Bogotá, and from there to Miami and then Phoenix for FDG metabolic and amyloid imaging with florbetapir (see NYT coverage). These people will travel to Phoenix in small groups to get PET studies going until a cyclotron that is currently under construction near Medellin can start providing labeled ligand for a local PET scanner that began operating in October 2011. “This travel is logistically challenging, and the team in Medellin is absolutely amazing in coordinating it,” Fleisher said.
All the above measures are also being taken in a much smaller group of relatives already affected with mild cognitive impairment or AD. The goal is to take sufficient biomarker measurements to pinpoint the earliest divergence between carrier and non-carrier for each of them, trace them forward into symptomatic AD, and integrate this information into a staged natural history of this form of Alzheimer’s. This information can then serve as a foundation for treatment trials, first in this population, but also, together with similar data from DIAN longitudinal biomarker studies of ApoE4 cohorts and ADNI and AIBL cohorts, for prevention trials in late-onset AD (LOAD). “Ultimately, we want to use treatment trials in early onset AD as models for late-onset AD,” Fleisher said.
What are the results so far? The data for the children and adolescents are not available yet. But as shown at conferences, data for people in their twenties are trickling in, and they show functional and even subtle structural brain changes that appear to precede amyloid deposition. Specifically, carriers had abnormalities compared to their non-carrying siblings and cousins in their brain activation patterns when they performed an established fMRI task asking them to associate and subsequently remember face-name pairs (Sperling et al., 2001). Carriers performed the task as well as non-carriers, but in doing so, they activated their hippocampi more strongly and deactivated their precuneus brain area less strongly. This is essentially the same pattern of change as previously reported for the later preclinical stages of other forms of early onset AD and, indeed, late-onset AD. Quiroz and colleagues presented these data at the Alzheimer’s Association International Conference (AAIC) in Paris in July 2011.
Also at this conference, Fleisher and colleagues presented a poster suggesting that this same group of twenty-somethings already have subtle morphological changes—meaning atrophy—in their brains. In a whole-brain comparison of gray matter volume between carriers and non-carriers, the 20 carriers had less gray matter in their temporoparietal and parahippocampal brain areas than 24 non-carriers who were otherwise matched in age, sex, education, and cognitive test scores. It’s well established that atrophy accelerates three to five years before dementia onset (e.g., Ridha et al., 2006). In this earlier work, the new signature may not have come up because the group was smaller, the imaging was not generally done in people this young, and what was done used more global measures of how the boundaries of regions of interest shift. The new API research uses voxel-by-voxel comparisons independent of regions of interest in people twenty years younger than their expected age at onset.
MRI offers a growing number of increasingly sensitive measures for AD research, and the API team put one more to the test. In a cohort of 18 mutation carriers and 22 controls in their thirties to early forties, Quiroz and colleagues worked with Brad Dickerson at Massachusetts General Hospital, Boston, to look for the cortical thinning signature Dickerson had developed in four previous studies in mild LOAD, MCI conversion to AD, and cognitively normal people who have amyloid and are being followed longitudinally. Dickerson had pinpointed nine regions of interest per hemisphere and found that atrophy, as measured by a thinner cortex in those regions, predicted that a cognitively normal person would develop dementia some eight years prior (Dickerson et al., 2011).
This is the first study of cortical thinning in the API population. In this cohort, mutation carriers on average had a 4.75 percent thinner cortex in these regions, Quiroz reported at AAIC in Paris. Most shrunken, by 6 to 8 percent, were the angular gyrus, the superior parietal lobule, and the precuneus regions. All nine regions showed a trend in the same direction, though not all are statistically significant, Quiroz said. Consistent with previous studies in other populations, these results point to neurodegeneration well underway by this stage, which in this population corresponds to what is generally called pre-MCI. With the Paisa AD mutation, affected carriers generally meet MCI criteria by age 44, a bit older than this cohort. In neuropsychological testing, this cohort, whose average age was 38, performed similarly overall to the non-carriers, though trends toward subtle decrements in word recall, verbal fluency, and recall of drawings were apparent. The earliest known cognitive deficit clearly demonstrated in this form of AD—in carriers in their thirties just like the ones studied for cortical thinning—is the visual binding memory deficit reported by Mario Parra and colleagues (see ARF related news story; Acosta-Baena et al., 2011 and Parra et al., 2010).
How do all these findings on brain imaging relate to Aβ? Amyloid PET results from API are unavailable as yet, but the first CSF and plasma data are beginning to roll in. At AAIC, Reiman presented the first cut, on 10 carriers and 10 non-carriers in the 18-26 age bracket. At this age, cognitive tests detect no difference, but brain function and structure measurements do. So far, Reiman reported, it looks as if carriers have elevated plasma Aβ42 but not Aβ40, suggesting that the presenilin 1 E280A mutation raises systemic absolute levels of this more aggregation-prone form of the peptide, as well as the Aβ42/40 ratio. (To some audience members, this finding hinted that middle-age elevated plasma Aβ42 might prove to be a risk factor in the general population as well.)
In CSF at this age, Aβ42 but not Aβ40 is elevated as well in carriers over non-carriers, Reiman reported at AAIC. This is consistent with the DIAN’s prior finding of elevated Aβ42 in carriers of a variety of early onset AD mutations in their twenties (see ARF DIAN London story; see ARF DIAN Honolulu story). Scientists generally assume that this reflects overproduction of Aβ, implying elevated levels of the peptide in the brain at an age where there is no fibrillar amyloid deposition yet. Not everything fits neatly, though: The same study finds a paradoxical reduction of CSF tau in carriers at this young age, upwards of 20 years prior to dementia, Reiman noted at AAIC.
What does this mean? It’s too early to make a strong statement, and it’s not proven that this form of early onset AD models LOAD, both Fleisher and Reiman cautioned in separate conversations. “Even so, at present it looks as if the functional and structural brain changes precede fibrillar amyloid deposition,” Reiman said, noting that this would be consistent with published work on reduction on FDG PET or in mitochondrial glucose metabolism in young adult ApoE4 carriers. Some studies are beginning to hint that fibrillar amyloid deposition, as visible by PET, happens soon after CSF Aβ42 has begun to drop. It is tempting, then, to speculate that the early functional and structural changes that Quiroz, Fleishman, and colleagues see might be happening in a situation of years of elevated Aβ levels but prior to when the brain deposits and, presumably, sequesters. This could imply that fibrillar amyloid deposition is an attempt by the brain to mitigate damage to synapses from an overabundance of prefibrillar forms of AD, Reiman said.
Both API and DIAN are pressing to add both cross-sectional and longitudinal data so they can address at what ages CSF Aβ42 starts dropping and how all markers the studies are tracking fit together. “More data on larger numbers of volunteers will sort this out,” Fleisher said. In the process, the currently proposed staging diagrams of preclinical (e.g., Perrin et al., 2009; Jack et al., 2010; Weiner et al., 2010; Frisoni et al., 2010) may get updated as some curves change their shape and slope or even trade places.
The result will be a knowledge base on the natural history of AD as a foundation for better clinical trials. For now, the API scientists are planning a first clinical trial as outlined in its pending grant proposal to the NIA, provided they can secure an appropriate compound, funding, and regulatory and ethical approval. At CTAD, Tariot emphasized that this trial is designed without pre-formed assumptions on which biomarker patterns will prove to be good outcome markers. Instead, it is designed precisely to address this question. “We must be humble about what we know,” Tariot said at CTAD, noting that regulators had advised API in previous planning meetings that their first trial should use a cognitive endpoint and include many biomarker readouts as secondary endpoints in order to learn as much as possible about them. Because the field does not know which biomarkers will prove to be outcome measures and how they will behave in response to a drug, the current trial is primarily frequentist with some adaptive elements. “We lack sufficient natural history data to build the computer models for a true Bayesian trial, and we have to be agnostic about the ability of biomarkers to predict treatment response. This is why we are not ready to use a Bayesian model yet,” Tariot said.
The proposed API trial, then, would use a change in a composite cognition measure as the primary outcome, looking for a slower rate of decline on drug versus placebo. Jessica Langbaum at the Banner Institute and colleagues elsewhere are developing this measure (see ARF related news story). Because this change will emerge slowly, the trial needs to be large and long. As proposed, the trial would enroll 300 participants. Two hundred carriers would be randomized 1:1 to treatment or placebo so no one would have to find out his or her mutation status; 100 non-carriers would be on placebo. The trial would feature an interim analysis after two years, guided by rules that assume biomarkers will change before cognition does. If the trial shows a positive biomarker pattern and/or clinical trends, then it will continue to five years, long enough to learn whether favorable cognitive changes are detectable.
Overall, the Alzheimer’s research field went from thinking a few years ago that this is too out-of-the-box to multiple groups now doing the same thing. In particular, industry scientists previously pointed to the absence of a regulatory path (see ARF eFAD essays). That path is clearer now, and involvement and support on the part of regulators have been evident. “The feedback from the regulatory scientists to API and DIAN has been incredibly valuable,” Tariot told the audience at CTAD (see ARF related news story; ARF news story). With an emerging regulatory path, the patients, the protocols, the tools, and some biomarker data in hand, researchers know the fate of those initial trials at this point would seem to lie squarely in the hands of funders.—Gabrielle Strobel.
This is Part 2 of a three-part series. See also Part 1 and Part 3. Download a PDF of the entire series.
No Available Comments
As trialists are retooling in the face of disappointing results in Alzheimer’s disease (AD), researchers in frontotemporal degeneration (FTD) are learning from AD woes and gearing up to take a seat in the front row. This year’s 4th International Conference on Clinical Trials in Alzheimer's Disease, (CTAD), held 3-5 November 2011 in San Diego, California, included a session on preparing for treatment trials in FTD. Presenters made the case that trials in FTD may stand a better chance of succeeding than those in AD. As research has uncovered many molecular pathways common to FTD and related disorders ranging from the common (AD) to the rare amyotrophic lateral sclerosis (ALS), any treatments developed for FTD may have broad applications and energize the field of neurodegeneration as a whole.
The session at CTAD represented the third meeting of the fledging FTD Treatment Study Group (FTSG). Since its inception in 2010 the group has been trying to drum up interest from pharmaceutical companies in conducting FTD trials (see ARF related news story). The reason for holding the meeting at CTAD was to catch a “broader audience who might not be involved in the FTD field at the moment,” said David Knopman of the Mayo Clinic in Rochester, Minnesota. “We wanted to see if we could attract clinical trialists from other areas and pique their interest by highlighting some of the new developments in FTD.”
The meeting also marked the beginning of FTSG’s formal association with the Association for Frontotemporal Degeneration (AFTD), a not-for-profit advocacy group that will serve as FTSG’s home, at least initially. “Being adopted by AFTD will allow us to grow and raise funds to help foster FTD drug development,” said Adam Boxer, University of California, San Francisco, who leads the FTSG. The funds will be used, for example, to encourage companies to share compounds that they might have otherwise abandoned so that they can be tested in FTD animal models. The funds will also help educate regulatory agencies about FTD-specific issues, further develop clinical trial methods, and create a patient registry, explained Boxer.
One of the main developments in the FTD field is that the molecular biology of the disease has been cracked open. “When I started to work in this area six years ago, we just knew about tau, but there have been such advances in our knowledge since then. As far as the genetic basis of disease, I think we virtually solved it. There are still many questions about how the different genes and proteins interact and how they lead to neurodegeneration, but we know who the main players are,” said Ian Mackenzie of the University of British Columbia in Vancouver, Canada.
In the “old” days, FTD—which is actually a group of diseases—was defined according to clinical criteria. Most people with FTD have behavioral variant FTD (or bvFTD), which degrades a person’s social skills and emotions. The next major category is semantic dementia, which hollows out their language; people can speak but no longer know the meaning of words. On the other hand, people with progressive nonfluent aphasia (PNFA) cannot speak fluently even though they know the meaning of the words they are trying to say. Finally, corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) are characterized by muscle weakness, rigidity, and/or parkinsonian symptoms.
More recently, researchers have adopted a classification of FTD based on the types of proteins that aggregate in cells (Mackenzie et al., 2009; Cairns et al., 2007; see also ARF related news story). The proteins involved are thought to play a role in the disease process, and are thus obvious targets for therapies.
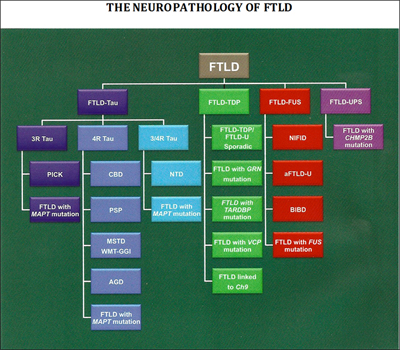
Researchers have uncovered many of the genes and proteins that play a role in FTD, leading to a new classification of disease based on its molecular pathology. Image credit: Nigel Cairns View larger image.
About 40 percent of FTD patients have intracellular accumulations of an abnormal form of the protein tau. This group of conditions, known as frontotemporal lobar degeneration (FTLD)-tau, includes most cases of PSP and CBD, as well as Pick’s disease. For PSP and CBD, the deposits consist almost exclusively of isoforms of tau with four microtubule-binding repeats (4R tau; Dickson et al., 2010), whereas in Pick’s disease, deposits contain 3R tau. Both 3R and 4R tau accumulate in neurofibrillary tangles in AD. In about 50 percent of FTD patients, including most who develop the disease in combination with ALS, the protein that accumulates is the TAR DNA binding protein 43 (TDP-43) (see ARF related news story). These patients fall into the FTLD-TDP-43 disease category. In 2009, investigators identified that the final subset of FTD patients (about 10 percent) have accumulation of the ALS-linked protein fused-in-sarcoma (FUS).
How do the clinical and protein-based classifications match up against one another? In the most common form of FTD, the behavioral variant, there is no clear correlation between clinical and molecular subtypes, which presents a challenge for selecting patients for trials. But with other subtypes, the relationship is clearer. Patients with the clinical symptoms of PSP and CBD almost always have pure tau pathology. As a result, patients with these conditions have been selected for early trials of tau-based therapies.
Why not test those therapies in Alzheimer's patients first? After all, neurofibrillary tangles have been a defining pathology of AD for a century. In reality, only a minority of AD patients have pure amyloid and tau pathology; most also have concurrent vascular or Lewy body disease. These other pathologies could mask any beneficial effects of drugs that target tau in those patients. “Clinical trials in FTD could be used for de-risking tau-related approaches to AD. A win here would rejuvenate the entire industry,” said Jeffrey Cummings, Cleveland Clinic, Nevada, at the meeting.
Another advantage of conducting trials in FTD is that in about 10 percent of patients, the disease is caused by a genetic mutation inherited in an autosomal-dominant manner (see ARF related news story). “We are talking about mutations that have a substantial impact on disease,” said Howard Feldman of Bristol-Myers Squibb, Wallingford, Connecticut, and the University of British Columbia in Vancouver. In his presentation at CTAD, Feldman suggested that genetic patients are obvious candidates for FTD trials. For years, AD trials focused on sporadic disease and excluded people with autosomal-dominant disease, but now some researchers are rethinking that strategy (see ARF related news story and ARF news story). “In FTD we have learned from the AD experience, and we are planning trials in at least one autosomal-dominant diagnosis, that is, FTD caused by progranulin haploinsufficiency,” Boxer told ARF. “Since the molecular defect is clear in this form of FTD, we may be able to hit an early home run that will help pave the way for wins in more common forms of FTD.” But others caution that this strategy is less straightforward than it sounds. “I agree that it needs to be done. But it will be challenging because the number of patients is so small,” Knopman told Alzforum.
Six genes have been associated with autosomal-dominant FTD. They are the MAPT gene on chromosome 17, which codes for tau; the PGRN gene, also on chromosome 17, which encodes the growth factor progranulin (see ARF related news story); the TARDBP gene on chromosome 1, which produces TDP-43; the VCP gene on chromosome 9, which codes for valosin-containing protein; the CHMP2B gene on chromosome 3, which expresses charged multivesicular body protein 2B (aka chromatin modifying protein 2B); and rare cases of FTLD are associated with mutations in the FUS gene (Van Langenhove et al., 2010 and Cairns and Ghoshal, 2010). Most recently, researchers identified an expanded GGGGCC hexanucleotide repeat in the noncoding region C9ORF72 as the cause of both chromosome 9p-linked FTD and ALS (see ARF related news story on Dejesus-Hernandez et al., 2011 and Renton et al., 2011).
These advances in genetics have highlighted the many similarities between FTD and ALS, and a number of researchers now view these two as opposite ends of a disease spectrum (see ARF related news story). But the overlap does not end there. FTD also shares common molecular pathways with Alzheimer’s. Beyond tau, FTD and AD may share molecular pathways that involve TDP-43 and progranulin, according to Mackenzie.
TDP-43 pathology is detected at postmortem in 25 to 50 percent of people with AD, and one study suggests that polymorphic variants in the gene, although they do not cause AD, increase the risk for AD; however, other studies have not found this association (see AlzGene). For its part, progranulin is an antagonist of the tumor necrosis factor α receptor, and has potent anti-inflammatory effects in animal models of arthritis (Tang et al., 2011). At CTAD, Mackenzie presented a model whereby progranulin may be involved in protecting the brain against damage. “It could be that reduced levels of progranulin put you at increased risk for neurodegeneration in general,” he told Alzforum. According to his model, FTD patients with progranulin mutations cannot protect their brain from age-related stress and damage, which eventually lead to FTD. AD patients have other primary mechanisms of toxicity, but having less progranulin leads to a worsening of symptoms in them, too. “The model is that if you develop AD and have reduced levels of progranulin, you will have increased neurodegeneration,” said Mackenzie. Although he acknowledged the proposal is highly speculative, he believes it is possible that progranulin-based therapies will work in both FTD and AD, as well as in inflammatory diseases.
In addition to these recent advances in understanding the molecular pathology of FTD, this past year has seen the development of clinical guidelines for the disease and the creation of two imaging projects à la ADNI. (Read Part 2 of this series for an update on those efforts.) “All the pieces are falling into place for clinical trials in FTD. We now need to convince industry to share promising drugs,” said Boxer.—Laura Bonetta.
This is Part 1 of a two-part series. See also Part 2. Download a PDF of the entire series.
No Available Comments
There are no treatments for frontotemporal degeneration (FTD), a common form of dementia in people younger than 65 years of age. That is one reason why speakers at a session titled “Clinical Trials in Frontotemporal Degeneration and Related Disorders” wanted to bring this somewhat neglected condition to the attention of the pharmaceutical industry and trialists working in Alzheimer’s disease (AD). The session was part of the 4th International Conference on Clinical Trials in Alzheimer's Disease (CTAD) held 3-5 November 2011 in San Diego, California.
In his presentation, Edward (Ted) Huey of Columbia University in New York City summarized the current state of affairs of FTD treatments. The few trials that have tested the approved therapies for AD in FTD patients have had disappointing results. “It appears that some of the patients who respond to cholinesterase inhibitors are those with AD pathology,” he said at the meeting. Memantine, too, has been tested in a small number of FTD trials. “So far it has been well tolerated in patients, but has not yet been shown to be efficacious,” said Huey. The results of the largest trial of memantine in FTD to date, conducted by Adam Boxer, University of California, San Francisco, will come out in 2012. “It should provide an answer,” said Huey.
Aside from therapies borrowed from AD, two FTD trials are testing compounds that target tau—a protein that accumulates both in some forms of FTD and in AD. Allon Therapeutics, headquartered in Vancouver, Canada, is sponsoring a Phase 2/3 randomized double-blind, placebo-controlled study to evaluate the safety and efficacy of davunetide in a type of FTD called progressive supranuclear palsy (PSP). Davunetide is an eight-amino acid peptide derived from a neuroprotective protein. It acts by maintaining and stabilizing the microtubular network (Divinski et al., 2004 and Divinski et al., 2006). Tau binds to microtubules and destabilizes them; davunetide presumably counteracts this effect, though its mechanism of action is not entirely clear. In PSP, the primary protein that accumulates in cells is a pathogenic isoform of tau, four-repeat tau. “There is little amyloid or other pathology, which makes the disease very attractive for a trial,” said Boxer, who is the study director (see ARF related news story).
Another company, Noscira, based in Madrid, Spain, is testing a different tau-busting agent in PSP. On 18 October 2011, the company announced that the last patient completed treatment in the yearlong Phase 2 efficacy trial of tideglusib, an inhibitor of glycogen synthase kinase 3-β (GSK-3β), one of the enzymes that phosphorylates tau. The trial tested two different doses of tideglusib (600 mg and 800 mg, taken orally once a day) versus a placebo in 146 patients with possible or probable mild to moderate PSP for 52 weeks at 24 sites in Europe and the U.S. “If these two trials demonstrate success with a tau-based drug in a pure tauopathy, this may help predict success in AD,” said Boxer.
Several presenters thought that targeting progranulin was a logical next step for FTD trials. Mutations in the progranulin gene may be the most frequent mutations among patients with autosomal-dominant FTD. The mutations lead to reduced levels of progranulin in blood and almost always produce TDP-43 inclusions in cells. Measuring progranulin blood levels in patients treated with a compound that boosts progranulin production may provide a way to monitor treatment effects (see ARF related news story). “A number of drugs that raise progranulin levels have been identified in high-throughput screens and tested in cell culture, and some have moved toward testing in animal models,” Boxer told ARF. “We hope that we will see clinical trials with some of these compounds next year.”
At the CTAD meeting, Michael Gold of Allon Therapeutics, which is sponsoring the davunetide trial, pointed out some of the regulatory advantages of conducting trials for orphan medications. Those are intended for the treatment of rare diseases that affect fewer than 200,000 people in the U.S., or that affect more but are not expected to recover the costs of developing and marketing a drug. (According to the presentation by David Knopman of the Mayo Clinic in Rochester, Minnesota, 20 people for every 100,000 aged 45 to 65 have FTD, and FTD has orphan disease designation.) “The FDA has indicated that drug approval based on a single trial is possible for many conditions that qualify for orphan indication,” Gold said at the meeting. According to the Orphan Drug Act, the sponsor of a clinical trial on an orphan disease can readily obtain guidance from the FDA along the way and market exclusivity for the drug being tested can be extended. Other advantages of conducting trials in FTD are that patients are younger and more enthusiastic to participate in trials than AD patients, Gold said. In addition, FTD patients’ symptoms are not complicated by changes due to aging, and the more rapid progression of disease makes trials shorter and requires fewer patients.
On the other hand, one challenge of conducting a trial with PSP patients is that the total number of patients with the disease is small, requiring that a given trial engages many different clinical sites, each of which recruits but a few patients. In the past, more sites have at times meant more sources for variability and error. “The FDA is responding to this problem and has released guidance on remote trial monitoring that may relieve that burden,” Gold said in his presentation. The draft guidance was released on 1 September 2011.
In addition to a favorable regulatory environment, new clinical diagnostic criteria for FTD, released earlier this year, may make trials easier to carry out. “Having clinical diagnostic criteria is critical to conducting clinical trials. If there is significant disagreement, as there is in the field of vascular dementia, for example, that scares away regulators,” said David Knopman of the Mayo Clinic in Rochester, Minnesota. “Although refinements are needed, there is broad agreement that the new criteria are good and valid, and people are comfortable using them.” One set of criteria is for behavioral variant FTD (bvFTD), the largest clinical subgroup of the disease (Rascovsky, et al., 2011). The second set of criteria is for the primary progressive aphasia (PPA) subtype of FTD (Gorno-Tempini, et al., 2011).
Another advance in the field has been the development of outcome measures that are appropriate for FTD. “We have to think outside the ADAS-Cog box,” Joel Kramer of the University of California, San Francisco, told Alzforum. “Up until recently, all available instruments were very memory focused, but those are not helpful for many types of FTD.” In his CTAD presentation, Kramer discussed tools his group has developed for measuring changes in executive function. One measure dubbed EXAMINER, for EXecutive Abilities: Measures and Instruments for Neurobehavioral Evaluation and Research, consists of a modular battery of tests that takes 30 to 40 minutes to administer and yields a composite score. Such tests may also be helpful in AD, Kramer believes. In the earliest stages of AD, memory is not sufficiently impaired for most current tests to capture changes, whereas it may be possible to detect shifts in executive function. His group has also made headway in developing measures for social behavior and emotion.
“The first step is to get pharmaceutical companies excited to conduct trials in FTD,” Kramer told ARF. “Once we have enough trials, we can build a consensus for outcome measures. Traditionally, the measures that are used in the first five to six trials become the gold standard.”
Another outcome measure that will be useful for FTD trials consists of changes in brain volume and function obtained through imaging. Taking the example of the landmark Alzheimer's Disease Neuroimaging Initiative (ADNI; see ARF related news story and ARF news story), two FTD-focused imaging studies have gotten off the ground. Howard Rosen of the University of California, San Francisco, described preliminary results from the FTLD Neuroimaging Initiative (FTLDNI). Funded in late 2009 by the National Institute on Aging and the National Institute of Neurological Disorders and Stroke, the project is following 120 FTLD patients with bvFTD, semantic dementia, and progressive nonfluent aphasia (PNFA), plus 75 controls. They are being examined at baseline, and then at six, 12, and 18 months using positron emission tomography (PET) and structural magnetic resonance imaging (MRI), as well as diffusion tensor imaging (see ARF related news story). “The changes we see in FTD are different from changes we see in Alzheimer’s,” Rosen said at CTAD. “And different variants of FTD are associated with different types of changes.” He presented data from one patient who showed a visible decline volume in both left and right lobes as detected by MRIs taken six months apart. “We will have easier time than ADNI in delineating changes because disease progression is faster,” said Rosen.
The second imaging initiative, headed by Boxer, is the Four Repeat Tauopathy Neuroimaging Initiative (4RTNI). Taking advantage of the FTLDNI infrastructure already in place at UCSF, this study will recruit 40 corticobasal degeneration (CBD) and 40 PSP patients, and study each for a year using a combination of clinical measurements, biomarkers, and 3T MRI scans. Both imaging projects have adopted standards developed by ADNI to enable comparisons.
One reason for conducting these studies is to find biomarkers specific for FTD. “One thing that is really missing is a biomarker approach that would enable clinicians to subdivide bvFTD into different entities according to molecular pathology,” said Knopman. It is not surprising that current trials are being conducted in patients with PSP, a type of FTD that is relatively uniform in terms of molecular pathology.
“It is difficult to tell the underlying pathology of FTD based on the clinical group, except for rare disorders,” said William Hu of Emory University, Atlanta, Georgia, speaking at CTAD. “The clinical syndrome is useful for tracking progression of disease, but it is hard to say who has FTLD-TDP-43 and who has FTLD-Tau based on clinical criteria.” To that end, his group is looking for sets of analytes that can differentiate between the two groups of pathologies.
With the molecular biology delineated, diagnostic guidelines and outcome measures developed, and longitudinal imaging studies underway, biomarkers are the last piece of the puzzle for a broad-based effort in FTD clinical trials. But even without such biomarkers, conducting clinical trials in certain types of FTD is already feasible. “The disease is not that rare, and there are enough patients out there who should make [the investment] worthwhile for pharmaceutical companies,” said Kramer. “Plus, there are incredible applications for AD and other diseases.”—Laura Bonetta.
This is Part 2 of a two-part series. See also Part 1. Download a PDF of the entire series.
No Available Comments
{kind=link}
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.