So you think Alzheimer’s is a neuronal disease? Well, yes, but you might as well think again. Even as the amyloid hypothesis is growing stronger with human genetics and longitudinal biomarker studies, decades of neurocentric research have given way to a broader appreciation that Aβ and tau intersect with the glial and vascular system and are subject to new forms of genetic regulation in the aging brain. What’s more, parabiosis shows that the blood itself carries signals into the brain that can powerfully influence its aging and degeneration. These topics dominated the first Zilkha Symposium on Alzheimer’s Disease and Related Disorders, hosted on April 4 by the University of Southern California. Gabrielle Strobel’s series covers the meeting highlights.
It’s Not All About You, Neurons. Glia, Blood, Arteries Shine at Symposium
On April 4, 150 researchers gathered in Los Angeles at the University of Southern California’s Keck School of Medicine for the first Zilkha Symposium on Alzheimer’s Disease and Related Disorders. The conference aimed to put a growing neuroscience institute on the map as a regional research powerhouse while celebrating a generous donor. Unlike many events meant to feature an institution, the program was jam-packed with new data from researchers from around the United States, Europe, and USC. Together, they articulated the latest thinking around the theme of how cells other than neurons, genes other than APP or presenilin, and proteins other than Aβ might conspire to bring on Alzheimer’s. The discussions reflected how the field’s collective effort to understand neurodegeneration is subtly shifting away from two decades of neurocentric research and toward the brain’s glial and vascular cells—even cells and soluble factors from the blood. “Not everything that happens in the brain is about the neurons. Many systemic influences reach the brain, and the brain is the end organ that reacts,” said Berislav Zlokovic of USC. Zlokovic co-organized the meeting with Rudy Tanzi of Massachusetts General Hospital in Charlestown, Massachusetts, and David Holtzman of Washington University, St. Louis.
The meeting’s name is a nod to Selim Zilkha, the 87-year-old patron of USC’s Neurogenetic Institute in his name, which hosted the conference. An Iraqi-born businessman, Zilkha has thus far given $30 million to the institute and will receive an inaugural philanthropy prize in July at the Alzheimer’s Association International Conference in Copenhagen, Denmark. “I have started many businesses in my life, but my most important legacy is this institute. The quest is to find a way to prevent or arrest Alzheimer’s disease. It is not important which individual or institution succeeds, but it is critical that someone does,” Zilkha told the audience.
Titled “Breaking through Barriers: Neuronal, Glial and Vascular Contributions,” the conference focused on how vascular, perivascular, and radial, astro-, and microglial cells contribute to neurodegeneration. Alzheimer’s genetics discoveries have spurred molecular biology studies of the neurovascular unit. The meeting combined new insights in how genetic factors such as Trem2 or microRNA, as well as environmental risk factors such as mid-life hypertension, sleep, or aging, affect the cells in these systems.
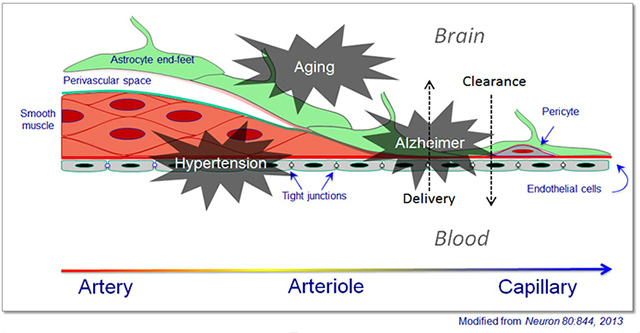
No one doubts that the brain’s vasculature contributes to dementia, but a precise definition of its role, specifically in Alzheimer’s and other neurodegenerative diseases, is only beginning to come into focus. Both subtle dysfunction and outright physical damage of the blood-brain barrier were the topic of several talks. Specifically, researchers are scrutinizing what happens at the neurovascular unit. This is where blood and neurons come closest to each other deep inside the brain, after the larger arteries entering from the top have arborized into small arterioles and capillaries. These delicate micro vessels consist of a single layer of endothelial cells enwrapped by a single layer of contractile cells called pericytes. Pericytes themselves are covered by the end-feet of astrocytes, glia that also stand in direct contact with neurons. This in essence, constitutes the blood-brain barrier.
This diagram of a neurovascular unit shows how the cellular constituents of the walls of incoming arteries change as they dive deep into the brain and arborize to become arterioles, then capillaries.
Epidemiologic research has shown that high blood pressure in a person’s 40s, 50s, and 60s increases his or her risk for dementia and Alzheimer’s in late life. Why is that? Over the past five years, Costantino Iadecola of Weill Cornell Medical Center, New York, has been building a case that hypertension slowly disables the brain’s micro vessels, rendering them unfit to adjust blood flow to suit the brain’s needs. On the one hand, this cerebrovascular dysfunction exposes the brain to hypertension pressing in from the periphery, raising the risk of stroke. On the other hand, it impairs the brain’s ability to locally increase perfusion where the brain is most active, leading to cognitive decline.
“Hypertension is the curse of the brain,” Iadecola said, “Reduced cerebrovascular blood flow is directly related to mortality—it is as if the brain is running out of breath.” In a January 2014 Alzforum Webinar, Iadecola outlined the field’s current understanding of how immune factors and oxidative stress damage the neurovascular unit to reduce cerebral blood flow.
At the Zilkha conference, Iadecola focused squarely on the pathogenic mechanism of hypertension. Even a nudge up in systolic pressure—from 120 to 140 mmHg—has been reported to impair cognition (Knecht et al., 2008). Therefore, early consequences triggered during a person’s mid-life pre-hypertensive state need to be simulated in animals to understand the underlying mechanisms.
Slow-Pressor Model
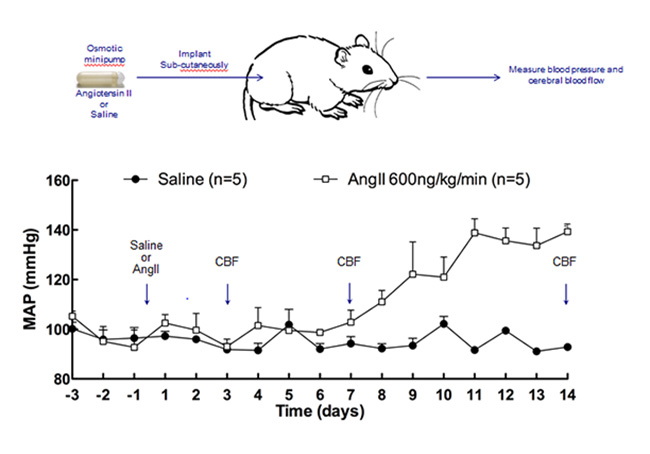
Iadecola’s lab approximates this pre-hypertensive state by delivering the angiotensin II peptide hormone through mini-pumps implanted into mice. Small daily doses increase arterial pressure slowly, starting at one week. The scientists measure cerebral blood flow through a cranial window above the whisker barrel cortex. This allows them to increase local blood flow directly by applying vasodilators through the window, or indirectly by stimulating a given whisker. This so-called slow-pressor model of mild hypertension showed that cerebrovascular function in the brain becomes impaired in response to angiotensin even before arterial blood pressure has started to rise (Capone et al., 2011).
Repeated squirts of the hormone angiotensin II gradually nudge up blood pressure in an attempt to model the effects of mid-life hypertension on the brain. Arrows indicate time points when changes in cerebral blood flow are being measured. [Image courtesy of Costantino Iadecola.]
In new data, the electron microscope shows physical changes to the endothelial cells that line the brain’s blood vessels, including an increase in vesicular transport and permeability to low-molecular-weight dextran. The blood-brain barrier is compromised, and labeled angiotensin II penetrates through the vascular wall into pericytes and astrocytic processes. In behavioral assays commonly used in Alzheimer’s research, the slow-pressor mice underperform. “This suggest that even in the pre-hypertensive state, you might already have vascular damage in your brain,” Iadecola told the audience.
What mediates this early damage? At the conference, Iadecola showed a series of unpublished experiments pointing an accusing finger at the perivascular macrophage, a type of cell that patrols the spaces just outside the blood vessel. This macrophage binds angiotensin II with the angiotensin receptor on its surface and, in response, spews radical oxygen species. Three lines of experimentation to manipulate perivascular macrophages—pharmacological or genetic deletion, and bone marrow transplantation with macrophages lacking angiotensin II receptors—suggested that without these cells, slow-pressor angiotensin hypertension no longer caused cerebrovascular dysfunction.
“This work shows that hypertension damages endothelial cells and pericytes early on. It suggests hypertension could deteriorate your cerebral blood vessels very slowly, for some 20 to 40 years. That makes complete sense to me,” Zlokovic said.
Perhaps no talk at an Alzheimer’s conference is complete without an experiment connecting the pathway at hand to Aβ or tau, and Iadecola’s was no exception. He showed that inducing a pre-hypertensive state in Tg2576 mice brought on amyloid deposition in their micro vessels at an age when they would not ordinarily form plaques yet. “You give these mice angiotensin, their blood pressure goes up, and then they make more amyloid plaques,” Iadecola said. This finding echoes a recent human study that found elevated amyloid deposition in some people with hypertension (Rodrigue et al., 2013).
The larger implication is that treating midlife hypertension would protect against dementia. This was partially borne out in 2013, when several epidemiological studies reported that the incidence of dementia was edging down in several developed nations, and attributed this in part to better management of cardiovascular risk factors such as hypertension (see May 2013 news story, Jul 2013 news story).
Managing blood pressure as people age is not straightforward, however. That’s partly because high blood pressure is a risk factor in mid-life, whereas old people generally fare better on slightly elevated rather than low blood pressure. Numerous treatment studies have shown benefits for ACE inhibitors or beta-blockers, but results vary. “It is pretty clear that midlife hypertension should be controlled, but we have no strong convergent evidence to know which treatment is best,” said Helena Chang Chui, who heads the department of Neurology at USC. For more on the blood-brain barrier, see part two of this series.—Gabrielle Strobel
Fluid Markers and Imaging Back Idea of Breached Blood-Brain Barrier
Besides hypertension (see part one of this series), a second source of neurovascular damage in the aging human brain took center stage at the inaugural Zilkha Symposium on Alzheimer’s Disease and Related Disorders, held April 4 at the University of Southern California, Los Angeles. This is the leakage of toxic serum proteins into the parenchyma due to a breach of the blood-brain barrier. Whether this is central to Alzheimer’s disease has long been controversial among scientists. About a dozen published studies have documented damage to the vascular system in postmortem Alzheimer’s brain; however, it is unclear what happens to the neurovascular unit years earlier, and whether changes there are an early driver or a late casualty of the disease process.
“I believe toxic blood-derived factors are an important cause of neuronal dysfunction. To really show that, we must develop molecular and imaging biomarkers that detect malfunction of the barrier and then embed these markers in cohort studies of early disease,” Berislav Zlokovic of USC told Alzforum. Zlokovic co-organized the Zilkha symposium.
Some of that biomarker development is starting to happen. In collaboration with the Alzheimer’s Disease Research Center at Washington University, St. Louis, USC researchers have tested a panel of vascular injury and repair markers in some 100 cerebrospinal fluid samples. The panel includes indicators of endothelial and pericyte damage, as well as angiogenic growth factors expressed in response. This panel shows changes in CSF early on, before tau and other markers of neurodegeneration or inflammation rise, Zlokovic told the audience. “We think that vascular factors can come into the brain silently and destroy it even before there is inflammation and neuronal injury,” he said.
Likewise, brain imaging is beginning to visualize effects of possible blood-brain barrier damage in early AD. For example, a pilot MRI study of cognitively normal older people at USC is showing areas of leakage, particularly in the hippocampus, Zlokovic said. Called dynamic contrast-enhanced (or DCE) MRI, the technique quantifies passage of a contrast agent. DCE MRI is used routinely to image the vasculature of tumors, but it can be adapted to measure the integrity of the cerebrovascular system itself. A 15-minute DCE sequence detecting local areas where the BBB is compromised could be added to the MRI scan already being done in existing human studies, said Art Toga. In May 2013, USC lured Toga, Paul Thompson, plus nearly 100 associated scientists away from the University of California, Los Angeles, in a spectacular cross-town academic raid. At USC, Toga now directs the new Institute for Neuroimaging and Informatics. INI encompasses the Laboratory of Neuro Imaging that Alzheimer’s researchers already know as LONI, as well as the Imaging Genetics Center (IGC), which hosts the imaging genetics consortium ENIGMA headed by Thompson.
In looking at the DCE MRI images, Zlokovic noticed leakage of blood into the hippocampus, that is, gray matter key to learning and memory. He was surprised because white matter was the primary site of BBB lesion in a separate USC imaging study. This study used mice which modeled the age-related loss of pericytes in the brain’s capillary walls that is known from AD postmortem studies. In the mice that lost pericytes, projections going from one side of the brain to the other were degenerating, suggesting functional disconnection.
So which one is most affected, gray matter or white? It could be both, Zlokovic said. The mouse study might show later consequences of a slow, chronic blood-brain barrier injury, in which local dysfunction precedes disruption of white-matter tracts. “Vascular changes basically intoxicate the brain and lead to subsequent losses in connectivity,” Zlokovic said. Both studies need more samples and independent replication to establish a sequence of events in the natural history of Alzheimer’s disease. “It will be very important to reproduce our imaging studies here and in aging and observational studies at other sites,” Zlokovic said.
The imaging data in the pericyte mice are the latest obtained from a previously published model of Alzheimer’s disease. The model simulates a two-hit hypothesis of Alzheimer’s, whereby age-related vascular damage interacts with Aβ to cause neurodegeneration. The researchers bred human mutant APP transgenics to PDGF receptor-β knockout mice. The crosses gradually lose pericytes with age, and as this happens, their blood flow in the brain slows down while their Aβ levels go up. Healthy pericytes ingest and degrade toxic proteins from the brain parenchyma; in the aging brain they can become overwhelmed and die. As they disappear, the wall of the micro-vessels thins even further, making it prone to blood components leaking out.
Recently, Abhay Sagare and others in Zlokovic’s group reported that depleting pericytes in APP transgenic mice worsened their phenotype. Pericyte loss drove up the mice’s brain and interstitial fluid Aβ levels, reduced clearance, and sped up amyloid deposition in the parenchyma and blood vessels. This induced pathological changes in tau and neuronal loss in the cortex and hippocampus, both of which APP transgenics themselves do not develop. “The blood-brain barrier breakdown makes these mice more complete models of AD,” Zlokovic said (Sagare et al., 2013; Bell et al., 2010).
The molecular mechanisms for this are under intense investigation, spurred in part by Alzheimer genetics findings. For example, ApoE4, besides being implicated in Aβ deposition, also has been shown to slow its degradation. Made primarily in astrocytes and released from their end-feet, ApoE may act through the LRP1 receptor on pericytes to suppress cyclophilin A signaling and thus keep a lid on the pro-inflammatory NFκB/MMP9 axis in pericytes. ApoE4 is less efficient at that than ApoE2 or ApoE3, in effect allowing a pro-inflammatory pathway to constantly burn on low heat in the vessel wall (May 2012 news story).
In new data presented at the Zilkha Symposium, Zlokovic noted that the same LRP1 receptor on endothelial cells interacts with the protein encoded by PICALM, an AD risk gene that came out of genome-wide association studies. PICALM is highly expressed in the brain’s blood vessels, Zlokovic said. There, it mediates clathrin-dependent endocytosis of Aβ bound to LRP1 into endothelial cells, and it also directs Aβ’s subsequent transcytosis across those cells to aid its drainage via the blood-brain barrier.
Broadly speaking in Alzheimer’s genetics, the GWAS era is nearing its end. Teased by some scientists as “God, What Awful Science,” genome-wide association studies have been controversial for doing little more than merely pegging genes to AD at high cost. Increasingly, sequencing of exomes or the entire genome of large groups of people is taking over in an effort to finger the actual pathogenic variants in the genes GWAS have flagged. With that, the real work begins, Rudy Tanzi of Massachusetts General Hospital, Charlestown, told the Zilkha audience. One such whole-genome sequencing project, of more than 1,500 samples, has already—even on preliminary analysis—yielded some 100 new mutations. This dataset predicts new functional variants in confirmed AD genes, including presenilins 1 and 2, as well as ApoE, clusterin, and Trem2, Tanzi said while previewing the WGS data.
For all their shortcomings, GWAS did confirm a serviceable list of genes as being implicated in the disease process. The list startled researchers when they saw just how many innate immunity, glial, and vascular genes it included. Together with the discovery of the microglial gene Trem2 by exome sequencing, the GWAS list re-energized neuroinflammation and -vascular research across the board. “We are getting very interested in how some GWAS hits affect the brain’s vascular system,” said Zlokovic. Terrence Town of USC said, “In the mid ’90s, everyone thought inflammation was an epiphenomenon in Alzheimer’s. Look how far we have come. We have a lot of data saying inflammation is deleterious in AD, but there are also good forms of inflammation.”
Trem2 has drawn particular interest in the past two years. This gene is highly expressed in microglia, which patrol the parenchyma in the immediate vicinity of the neurovascular unit. Recently, a genetic imaging study showed that Alzheimer's Disease Neuroimaging Initiative participants who carry a Trem2 pathogenic variant lost brain tissue much faster than non-carriers. “We see a dramatic effect size for Trem2,” USC’s Thompson told the Zilkha symposium audience (Oct 2013 story on Rajagopalan et al., 2013). For more on microglia in the aging brain, see part three of this series.—Gabrielle Strobel
In Revival of Parabiosis, Young Blood Rejuvenates Aging Microglia, Cognition
As the brain ages, its microglial cells turn sluggish in their task of ingesting and degrading toxic products, and the flow of blood through its micro vessels slows. Are there components in the blood that age the brain—and can renew it? At the Zilkha Symposium on Alzheimer’s Disease and Related Disorders, held April 4 at the University of Southern California, Los Angeles, Tony Wyss-Coray of Stanford University, Palo Alto, shared unpublished results from an ongoing study that first characterized a microglial aging phenotype and then partially reversed it with still-unknown factors from the body’s systemic milieu.
The data offered a provocative new twist on the old specter of rejuvenation with young blood. It also reflected the power of heterochronic parabiosis, a surgical protocol of conjoining the blood supply of a young and an old mouse to study complex pathophysiological processes. The new data presented at the Zilkha symposium receives support from a flurry of separate parabiosis papers published on May 4. Led by scientists at the University of California, San Francisco, Harvard Medical School, and other institutions, these three papers demonstrate striking benefits of young blood on cognitive function, synaptic plasticity, neurogenesis, and the cerebral vasculature of old mice. The beneficial effects are not limited to the brain, and the operative factors can be identified, as one study reports that growth differentiation factor GDF11 empowers young blood to bestow regenerative oomph to old muscle.
These two mice, one old one young, live with a shared blood supply. [Image courtesy of Tony Wyss-Coray.]
Aging brings with it not only a decline in cognition but also a smoldering inflammation within the innate immune system. “This low-grade chronic inflammation is bad news,” Terrence Town of the University of Southern California in Los Angeles said at the Zilkha conference. In the brain, this manifests as an abnormal state of that organ’s main resident immune cell, the microglia. For example, expression of the microglial activation marker CD68, a lysosomal protein, rises with age. Electron micrographs of aging brain show microglia with an enlarged, dense nucleus, shriveled Golgi cisternae, few lysosomes, and vesicles jammed with lipufuscin granules. “Aging microglia clearly look abnormal,” Wyss-Coray told the conference audience. They behave abnormally, too, hardly phagocytosing in culture when presented with their usual substrates.
To see whether this is an internal affair of the aging brain or influenced by the periphery, Wyss-Coray returned to a blood-sharing experiment called parabiosis. His lab had previously used it to show that a young systemic environment can essentially rejuvenate neurogenesis and other aspects of the aging brain (see Nov 2009 news story, Mar 2013 news story).
Parabiosis involves suturing the body walls of two mice together such that their capillaries fuse. The mice then live like Siamese twins joined through their blood supply. At the Zilkha conference, Wyss-Coray said that pairing an 18-month-old with a 3-month-old mouse, and letting them live together for five weeks, reversed microglial aging. Microglial activation as measured by CD68 expression was down in the brains of old mice exposed to young blood. In the electron microscope, the old mice’s microglia looked like those of young mice, with a normal-sized, light nucleus, larger Golgi cisternae, more lysosomes, and less lipofuscin.
To the eyes of some scientists at the conference, the nucleus in microglia from old mice looked as if it contained dense chromatin that would allow less protein translation. Wyss-Coray replied that he does not know for sure how the ultrastructural changes in aging microglia relate to gene expression and function.
That said, his lab did compare the microglial transcriptome from old mice paired with other old mice to that from old mice paired with young mice. They saw that blood supplied by a young mouse did indeed largely reverse the gene expression phenotype of microglial aging. This includes age-related increases not only in CD68 and in the complement component C1qB; but also age-related decreases in progranulin, the transcription factor EGR1, and many other expression changes.
In a separate study, Ingenuity Pathway Analysis of gene expression profiles of the aging hippocampus pointed to a synaptic plasticity network anchored by the transcription factors Creb (cAMP response element-binding protein) and EGR1 as being most preserved in rejuvenated old mice. Golgi silver staining spotted more spines on the dendrites of old mice when each had each partnered with a young mouse. A paper published by Wyss-Coray and his former postdoc Saul Villeda and colleagues on May 4 in Nature Medicine reports additional findings to back up the claim of revitalized synapses in heterochronic old mice. These include strengthened long-term potentiation as seen with electrophysiology of cultured hippocampal slices, as well as mechanistic experiments using local expression of dominant-negative Creb and RNA interference of Creb to pinpoint this transcription factor as a hub in the requisite signaling network. “There is some sort of reactivation of a synaptic plasticity network in an old mouse exposed to young blood, ” Wyss-Coray said.
Whether these changes at the molecular and cellular level amount to better function is difficult to assess in parabiotic mice. The pairs run the rotarod together, but rigorous behavior assays are not possible. Instead, the Stanford scientists decided to model parabiosis by transferring young plasma into an old mouse once every three days for three weeks. In this study, old mice injected with plasma from young mice outperformed untreated old mice in the radial arm water maze and a fear-conditioning test. The treated mice also recapitulate other previously shown parabiosis phenotypes, including more neurogenesis, synaptic plasticity, spine density, and less neuroinflammation. The effects are not due to steroid hormones, and happen equally in male and female mice, Wyss-Coray said.
“These results are stunning. Extremely interesting,” commented Berislav Zlokovic of USC.
Are they too good to be true? “That is what some reviewers said,” Wyss-Coray replied dryly. The latest findings of this line of research are not yet published, but previous work appeared in 2009 and has been awaiting independent replication. Villeda has started his own lab at UCSF, where new students have reproduced the findings. Beyond that, few other labs have independently replicated them yet.
That may be beginning to change with a new paper, released also on May 4, in Science magazine. In this study, researchers led by Lee Rubin at the Harvard Stem Cell Institute in Cambridge, Massachusetts, report that young blood reinvigorated blood vessels of the neurogenic niche in the brain of old mice. In heterochronic old mice, the volume of cerebral blood vessels almost doubled. They formed new branches and allowed more blood to flow through (see movie below). This, in turn, supported a robust increase in the number of neural stem cells in the old mice’s sub-ventricular zone, a brain area that is a source of new neurons throughout life but runs dry in aging (see news story on Science Now).
Blood flow in an old mouse brain. [Courtesy of John Chen and Greg Wojtkiewicz]
Blood flow in a rejuvenated old mouse brain. [Courtesy of John Chen and Greg Wojtkiewicz]
This paper confirms Wyss-Coray’s 2009 findings that factors in young blood stimulate neurogenesis in the old brain and that factors in old blood slow neurogenesis in the young brain. Rubin’s paper refines Wyss-Coray’s by reporting that it is not until mice are truly old—in this case 21 months—that their blood impairs neurogenesis of young mice. Blood from 15-month-old mice did not, suggesting that it is only during aging that these negative factors accumulate in the blood.
These new papers aside, why were scientists at large slow to catch on to the potential of parabiosis for the study of brain aging? It may be partly because parabiosis has fallen out of favor over the past two decades and appears only now to experience a small revival. Greek for “living alongside,” this experimental system has a storied history. Used widely in physiology and endocrinology research during the first 70 years of the 20th century, parabiosis advanced the fields of growth and sex hormones and set the stage for the discovery of parathyroid hypertensive factor. Parabiosis showed the presence of the satiety factor that was later called leptin, a discovery that garnered the 2010 Albert Lasker Basic Medical Research Award (Coleman 2010). In 1972, parabiosis showed that old rats lived longer and were more vigorous when conjoined to young rats (Ludwig et al., 1972. Incidentally, the “trans” in this citation stands for ‘Transactions,” not "Transylvania"). Alas, the “creep factor” of creating a surgical bondage in the service of science may have set animal care committees against the technique, Wyss-Coray said, and it faded from use.
In a recent review article arguing for a return of parabiosis to study the pathophysiology of age-related disease, Wyss-Coray writes that paired mice fare better than many other animal models exposed to pathogens, traumatic injuries, cancer, or debilitating mutations (Eggel and Wyss-Coray, 2014). In Los Angeles, he noted unpublished recovery data showing that the pairs resumed grooming and nesting, and lived a full lifespan.
Wyss-Coray’s lab initially learned parabiosis from fellow Stanford scientist Tom Rando, who used it to stimulate regeneration in liver and muscle (Conboy et al., 2005). Since then, heterochronic parabiosis has boosted recovery in models of multiple sclerosis and heart failure due to weakening cardiac muscle (Ruckh et al., 2012; Loffredo et al., 2013). Indeed, this last study, by the groups of Lee Rubin and Amy Wagers, also at the Harvard Stem Cell Institute, gave rise to the third paper published on May 4. In it, researchers led by Wagers report that the circulatory protein GDF11 rejuvenates not only heart but also skeletal muscle. The scientists first characterized how heterochronic parabiosis restored in old mice the muscle satellite cells that promote muscle healing, and then went on to show that daily injection of recombinant GDF11 generated much the same phenotype. The new paper by Rubin et al. on the effects of young blood on the neurovascular niche of aged mice also showed that daily injection of GDF11 alone pulled off about half the beneficial effect on the brain’s capillaries and neurogenesis as that noted for whole young blood in parabiosis.
In the view of other scientists at the Zilkha conference, the apparent success of the plasma transfer protocol and even targeted injection of candidate humoral factors is likely to prompt more laboratories to take up parabiosis.
In a press release issued by Harvard University, Rubin is quoted extensively. “We think an effect of GDF11 is the improved vascularity and blood flow, which is associated with increased neurogenesis. [This] should have more widespread effects on brain function,” he said. "We do think that, at least in principle, there will be a way to reverse some of the cognitive decline that takes place during aging. It isn't out of question that GDF11, or a drug developed from it, might be capable of slowing some of the cognitive defects associated with Alzheimer's disease."
Rubin is further quoted as saying that a future treatment for Alzheimer’s might be a combination of a therapeutic that reduces plaques and tangles with a potential cognition enhancer like GDF11.—Gabrielle Strobel
The new findings of Villeda et al. are remarkable and thought-provoking, as they suggest the possibility that transfusion of blood/plasma from young humans can restore cognitive function in patients with mild cognitive impairment or Alzheimer’s disease. Indeed, in a previous study it was reported that cognitive function was improved in Alzheimer’s disease patients who underwent plasma exchange (Boada et al., 2009). It will be critical to identify the presumptive factor(s) in the plasma of young animals that stimulate(s) neuroplasticity in the brains of old animals. Possibilities range from a neurotrophic factor to a protein that promotes removal of toxic molecules from the brain.
Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
The neurovascular focus of the first Zilkha Symposium on Alzheimer’s Disease and Related Disorders, held April 4 in Los Angeles, started off with talks on the blood-brain barrier and midlife hypertension (parts one and two of this series). Proper blood pressure is crucial not only to keep liquid moving inside the brain’s vessels, however. According to Maiken Nedergaard, University of Rochester, New York, arterial pressure also drags cerebrospinal fluid along the vessel’s outside, and this type of flow is equally important. Normally, the pulsatility of the brain’s arteries creates a convective force that draws CSF into the brain from the space around its artery walls and back out again along its veins. Gently propelled by this peristalsis, CSF penetrates deep inside the brain’s parenchyma. There, in the neighborhood of the neurovascular units, CSF exchanges with interstitial fluid, percolating through and around astroglial end-feet toward nearby veins, along which it then exits the brain. In so doing, it takes with it neuronal waste products, essentially rinsing neurons like so many pots and pans in a dishwasher (see March 2013 news story, Jan 2014 Alzforum Webinar, Wikipedia video below). “This is how the brain exports its waste,” Nedergaard said.
In Los Angeles, Nedergaard reported that this flow wanes as people get older. “We see a striking reduction of CSF influx into the depth of the brain with age,” Nedergaard said. For one, she said, new experiments show that the pulsatility of the small penetrating arteries weakens alarmingly. For another, the distribution of the aquaporin receptor changes. Normally, these receptors are located on the astrocytic end-feet facing the arteries, where they facilitate uptake of fluid from the peri-arterial space and drive glymphatic flow toward nearby veins. In old mice, the receptor occurs all over the astrocytes in a way that facilitates fluid retention inside this cell more than it aids directed flow from arteries to veins. This may be one way in which astrogliosis damages the aging brain, Nedergaard said.
It is well known that waste products such as Aβ and other aggregation-prone proteins are cleared not only alongside veins but also across the blood-brain barrier into the venous bloodstream (part two of this series). Even so, Nedergaard argued, Aβ and α-synuclein, being synaptic proteins, need an interstitial fluid flux to reach the BBB, whether they then get transported across or washed out by way of paravenous efflux.
Do We Sleep To Clear Our Minds?
Glymphatic clearance peaks in mice when they snooze. This is so not only because aquaporin receptors are more highly expressed in sleep, but also because the interstitial spaces themselves expand. “The interstitial volume decreases in wakefulness. It is a huge difference that would make a big impact on convection flux,” Nedergaard told the audience. Most of the molecular underpinnings of the glymphatic system remain to be understood; in the meantime, a good therapy for an aging brain may be to sleep more, Nedergaard said.
This, too, is a recommendation from a growing literature on sleep and neurodegenerative disease more broadly. It appears to be a bi-directional relationship. Sleep apnea or shift work in mid-life increases people’s risk for dementia, and once dementia has taken hold, it further alters sleep patterns, with frequent and active waking periods after midnight. Another example of this relationship may be REM sleep behavior disorder, which predisposes to Parkinson’s and dementia with Lewy bodies, both of which in turn count excessive daytime drowsiness among their symptoms.
In healthy adults, the Aβ concentration in the cerebrospinal fluid is subject to a circadian fluctuation that is somehow lost as people reach their 60s and 70s. David Holtzman of Washington University, St. Louis, tries to define how neural activity, the wakefulness hormone orexin, and sleep deprivation influence the concentration of Aβ and tau in those very interstitial spaces that get flushed by glymphatic flow.
NMR solution structure of orexin. Knocking out this hormone induces sleep and furthers glymphatic clearance of waste proteins, including those involved in Alzheimer’s.
Some years ago, Holtzman’s group showed that synaptic activity regulates Aβ levels in extracellular spaces, and that orexin increases Aβ levels in the interstitial fluid (Sep 2009 news story). At the Zilkha conference, Holtzman showed new data suggesting that orexin does not do this directly, by affecting neuronal signaling locally; it does so indirectly, via its influence on how much the mice sleep. Local orexin expression experiments by way of injecting lentiviral constructs into the brain indicated as much. So did orexin knockout mice crossed with APP/PS1 transgenic mice, which sleep more and have less soluble and deposited Aβ. Accordingly, other ways of changing the amount of sleep also influence the amount of Aβ in fluids; for example, certain sleep-inducing drugs may lower CSF Aβ in human volunteers, Holtzman said.
Tau, the other hallmark protein of Alzheimer’s disease, is becoming a target of such studies, as well. Tau is not generally thought to be related to sleep; however, evidence is growing that it is being released into the interstitial space—more so during periods of high neuronal activity, i.e., wakefulness. Recently, Holtzman’s lab showed that driving up excitatory activity increases ISF tau (Feb 2014 news story). In Los Angeles, Holtzman noted that the half-life of tau in the ISF is too long to expect the fast spike in response to sleep deprivation that has been seen with Aβ; however, chronic sleep deprivation might drive up tau. Other labs have begun linking sleep to tau in transgenic AD mice (De Meco et al. 2014; Rothman et al., 2013; Cantero et al., 2012).
What Have microRNAs Got to Do With it?
While some groups known for their work on the signature proteins of Alzheimer’s disease have started exploring physiological processes such as glymphatic clearance and sleep, other groups are venturing into the uncharted swaths of the human genome to better understand gene regulation in the disease. Bart de Strooper, KU Leuven, Belgium, is searching the non-coding tracts that make up the vast majority of human DNA for genetic elements that may be important in age-related neurodegeneration. “Do not call anything in the DNA ‘junk.’ We have to scrutinize all of it,” de Strooper said. In Los Angeles, he presented new data on the microRNA 132 in Alzheimer’s disease.
MicroRNA-132 precursor, secondary structure. [Wikimedia Commons image by permission of User:Ppgardne.]
MicroRNAs are small wisps of 18 to 23 nucleotides that bind to the 3’ UTR of target mRNAs and suppress their translation. They are encoded as independent genes, some in introns of other genes, others in what used to be dismissed as “junk” DNA. De Strooper’s lab started exploring microRNAs by deleting brain expression of dicer, an enzyme needed to process microRNAs. The resulting phenotype of tau phosphorylation, inflammation, and neuron loss intrigued the scientists sufficiently to look for microRNA binding sites in Alzheimer’s genes such as BACE and APP, where they indeed found them. Subsequent microRNA profiling of 49 AD brain samples from the Netherlands Brain Bank showed that expression of 41 microRNAs was different in the AD than control samples, and a follow-up study in AD brains from the MRC London Brain Bank confirmed the pattern.
Of those 41, miRNA-132 proved to be markedly downregulated in the AD hippocampus and prefrontal and temporal cortex, and more so the farther advanced the patient’s disease had been by Braak staging. Deep sequencing and additional techniques confirmed that this finding is robust and reproducible, De Strooper told the audience.
It’s not just Alzheimer’s. This microRNA is down in Huntington’s, progressive supranuclear palsy, frontotemporal lobar degeneration, and amyotrophic lateral sclerosis. Berislav Zlokovic of the University of Southern California noted that stroke studies have shown the same thing. “We see this remarkable miRNA-132 downregulation long before neuron loss, so we think it is important,” de Strooper said.
Sequence analysis predicts 407 miRNA-132 target genes in the human genome, including the genes for tau, GSK3β, and sirtuin1. In-vitro binding validated that, de Strooper said. To get a sense of what miRNA-132 might do, the scientists knocked it down it in zebrafish, which then had trouble swimming. In the developing zebrafish brain, the little RNA is strongly expressed in radial glia. These are progenitor cells that also guide migrating neurons. At the molecular level, miRNA-132 acts via notch, a γ-secretase substrate that is well known to be important in glial development. The microRNA affects notch by binding directly to the mRNA of the transcription factor CTBP2, which connects to notch via sirtuin-1.
Much remains to be found in this emerging pathway. “Even so, our data point in a speculative way toward a role of neurogenesis in brain aging, maybe some protective role in AD,” de Strooper said.
In the discussion, de Strooper said that the world of RNA should be explored with an emphasis on normal and diseased human brain for the time being, noting that the prospect of miRNA-based therapeutics appears premature.
The field’s growing interest in new cellular and molecular players in the pathogenesis of AD does not mean the original ones—APP and presenilin 1 and 2—have been set aside. At the Zilkha symposium, researchers presented new findings and model systems. Sam Sisodia of the University of Chicago discussed recently published research suggesting that Aβ generation in cells other than excitatory neurons is important in Alzheimer’s pathogenesis (see Veeraraghavalu et al., 2014).
Rudy Tanzi of Massachusetts General Hospital, Charlestown, introduced a way of growing human neuronal precursor cells that express familial AD mutations of those genes in three dimensions by using a matrigel mixture; this is based on the work of Doo Yeon Kim of MGH. As the cells secrete Aβ, the gelatinous consistency of the substrate restricts the peptide from diffusing away, and the cultures form both Aβ oligomers and amyloid plaques. Intriguingly, they also form hyperphosphorylated tau and even neurofibrillary tangles that stain with conventional silver stains, Tanzi told the audience. Beta- or γ-secretase inhibitors prevent the formation of both plaques and tangles in the cultures, whereas tau-based inhibitors prevent formation of tangles even in the continued presence of ample amyloid plaques, Tanzi said. This culture system might be useful for drug screening, and it could potentially recapitulate aspects of the pathology of other neurodegenerative diseases that feature secreted pathogenic proteins.—Gabrielle Strobel


 Mark Mattson
Mark Mattson 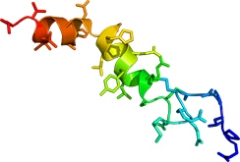

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.