German-International Alzheimer research strutted its stuff in Bavaria, the state where Alois Alzheimer was born. Scientists met at a secluded lake nestled deep in the Bavarian Alps in the southern part of the state, an area known for its bucolic landscape, skiing, and expressionist art.
Eibsee: 8th Gathering of German-International Alzheimer’s Researchers
As scientists around the world enjoy their holiday family retreat, a few of them may recall a recent gathering of a different sort. There has come to be an unmistakable family feeling to the annual Eibsee conference, where many of Germany’s leading Alzheimer disease researchers meet with a few invited international colleagues at a scenic little lake in southern Bavaria. Nestled in picturesque isolation at the foot of Germany’s tallest mountain, the Zugspitze, the hotel hosting this conference cocooned the 85 attendees into a meeting room, a restaurant, and a bar with a fireplace from October 29 to November 1, ensuring ample time for discussion, exchange of ideas, and occasional commiseration.
Organized annually by Christian Haass and his colleagues Regina Fluhrer and Andrea Dankwardt at Ludwig-Maximilians-University Muenchen, the Eibsee Meeting on Cellular Mechanisms of Alzheimer’s Disease brings together many of the grant recipients of the Collaborative Research Center 596. This is a funding mechanism through the Deutsche Forschungsgemeinschaft, the German equivalent of NIH funding. In his introductory remarks, Haass noted that this funding vehicle, which began in 2001 and undergoes international review every four years, has fostered collaboration across institutional boundaries at the various institutes pursuing neurodegeneration research in Munich, Germany’s historic center of that field, and among institutions in other cities. In addition, the Eibsee conference invites some of its international reviewers and other prominent AD scientists. This year, speakers included Todd Golde of the Mayo Clinic in Jacksonville, Florida, Bart de Strooper of the Flanders Institute for Biotechnology in Leuven, Belgium, Huilin Li of Brookhaven National Laboratory in Upton, New York, Robert Vassar of Northwestern University in Chicago, Illinois, Philip Wong of Johns Hopkins University in Baltimore, Maryland, Ganesh Shankar of Harvard Medical School, Guriqbal Basi of Elan Pharmaceuticals in South San Francisco, and Melanie Meyer-Luehmann of Massachusetts General Hospital in Boston.
Besides the science itself, discussion at meals and the bar revolved around life as a neurodegeneration scientist in Germany and Belgium these days. On the whole, European researchers had few complaints about the funding situation in their respective countries. Getting their data published is getting more difficult for some, however. Scientists talked about how they are spending increasing amounts of time contending with negative reviews and reformatting manuscripts for repeated submission as their papers get rejected. Besides cutting into research time, this slows the dissemination of new findings as even established groups can spend a year or more securing publication of a given study. The solution scientists advocated was for each reviewer to work toward a less rivalrous process. The technical standards in AD research have risen in the past decade, de Strooper said, adding that AD researchers as a group can support each other by balancing technical rigor with good faith and letting the field serve as the corrective. If a high-profile paper years later has generated no follow-up in the literature, it often means other labs were unable to replicate the finding. The sentiment was that overall, this is better than slowing publication of deserving manuscripts.
To quote one example, Golde presented an example of a recent study that is controversial and might have been rejected for publication in Nature. The paper reported that certain NSAIDs bind not the γ-secretase, as was assumed, but instead the Aβ region of APP itself (Kukar et al., 2008). Despite what Golde believes was multi-pronged evidence to support the main conclusion, he noted that one reviewer discouraged publication of the data. The editor overrode, and independent colleagues in several labs are now testing the paper’s claims. For one, Gerd Multhaup of the University of Berlin, reported at the Eibsee that by using a different technical approach, his group has also detected direct binding of such NSAIDs to the same Aβ sequence (see also Part 2 of this series). In time, the literature will show how the finding stood up.
A separate issue that had tongues wagging was a major initiative by the German government, announced in its raw outline this past spring, to create a new national research center dedicated to neurodegenerative diseases. To be called Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE), the center will be based in the provincial city of Bonn, the country’s former capital, with sites in Munich, Tuebingen, Goettingen, and the eastern cities of Magdeburg and Witten. To the surprise of many, more established centers for AD research such as Heidelberg, Hamburg, or Berlin, were not chosen as key nodes in this future research network. The goal is to integrate research across the domains of basic mechanisms, translational studies, and also patient care. The Alzforum will cover that effort in its own right as it gets underway.
This space will feature a series of summaries of the major themes, as well as selected highlights from the 30 talks and 18 posters at this conference. Meanwhile, the Eibsee gallery features shots of the setting, speakers, and more.—Gabrielle Strobel.
This is Part 1 of a seven-part series. See also Parts 2, 3, 4, 5, 6, 7.
In a wide-ranging lecture at the 8th Eibsee Meeting on Cellular Mechanisms of Alzheimer’s Disease, held October 29 to November 1 in Germany, Todd Golde of the Mayo Clinic in Jacksonville, Florida, subsumed summaries of new data from three research areas in his laboratory under a broader theme that is coming to the fore across the field. That is, recent clinical data and some animal research are converging into a realization that anti-amyloid treatments may only succeed if given preventively, not as treatments once a person has received a diagnosis. As one example from his own ongoing research, Golde mentioned a treatment study in mice that tried to assess what might be the best window of amyloid removal.
Golde started by noting that the amyloid hypothesis remains the basis of much mechanistic and translational research in the field. Besides the much-cited genetic and subsequent animal and cell culture work, less appreciated support for the amyloid hypothesis comes from related brain amyloidoses such as the British and Danish familial dementias. In these diseases, a completely different protein, when mutated, can be cleaved to release a peptide that deposits as amyloid, leads to tau pathology, and causes dementia. No one truly knows why neurons die in Alzheimer’s or related diseases, and yet many people in academia and industry are confident that blocking Aβ aggregation will eventually have therapeutic value, Golde said.
Golde estimated that the total Aβ deposited in Alzheimer disease equals some 10 to 20 micromoles of Aβ. Based on measurements of daily turnover, this represents some two to five years’ worth of brain Aβ production. By comparison, about 10 nanomoles of Aβ accumulate in the brain of a 21-month-old Tg2576 mouse, and this reflects about a month’s worth of its brain Aβ production. In essence, APP transgenic mice without neuronal loss really model preclinical AD, not the symptomatic disease, Golde said, echoing a point Karen Ashe has been making for some time. That, then, is what scores of mouse therapeutic studies have been testing over the years, and it may also partly explain why mouse studies on a wide range of therapeutics succeed relatively easily.
One familiar treatment option is to slow Aβ production with γ-secretase inhibitors, of which one candidate is in Phase 3 trials, and a new generation of more selective inhibitors is entering Phase 1 (see related SfN/ICAD story). To ask when and for how long a patient would have to take such an inhibitor, postdoctoral fellow Pritam Das and colleagues administered the experimental γ-secretase inhibitor LY-411575 to Tg2576 mice at different ages and for different periods of time. This is relevant because amyloid deposition is thought to go through an initial, slow seeding phase, and then grow exponentially for some time before reaching a fairly stable plateau. This plateau is seen in Tg2576 mice from the age of 20 months onward, and in people for much of their years with clinical AD. Das and colleagues treated the mice for defined periods of time, either from four to seven months of age, or from seven to 10 months, or from 12 to 15 months. When examined at 15 months for brain Aβ levels and plaque load, the mice revealed that the only treatment that truly worked was the first. In other words, giving a γ-secretase inhibitor around the time of seeding held off subsequent amyloid deposition even though both the APP transgene and γ-secretase remained active after seven months of age. But once the deposition train had left the station, the break on γ-secretase had little effect. “This means that once the nucleation of amyloid formation has occurred, it is hard to stop further aggregation with secretase inhibition. And it really supports prophylactic rather than therapeutic targeting of Aβ,” Golde said. Newer γ-secretase inhibitors are entering the clinic, so far mostly for safety and pharmacokinetic/pharmacodynamic and biomarker studies (see SfN story).
γ-secretase modulators (GSMs) represent a separate therapeutic avenue, and it had a crisis this past summer. That endeavor draws on a broad consensus that a safer approach than all-out γ-secretase inhibition can be found in tweaking the enzyme complex such that it merely shifts its output toward more of the shorter forms of Aβ and fewer of the longer forms, particularly the highly fibrillogenic Aβ42. At the scientific level, this idea is widely accepted, and researchers led by Golde, Eddie Koo, Sascha Weggen, and other groups have elaborated it with the discovery of a range of compounds that either lower or increase the selective production of Aβ42. But at the clinical level, a drug identified early on in this line of research failed utterly this spring (see ICAD story). Scientists ascribed this downer to the chosen compound, to the lack of accompanying CSF biomarker data, and to having tried treatment rather than prevention. Flurizan, they decided, had really not tested the underlying notion of γ-secretase modulation in the first place, because too little of it had reached its target in the human brain. One setback later, follow-up research on second-generation GSMs, coupled with more detailed dissection of underlying mechanisms, is moving to center stage. “This is not the kiss of death for GSMs. New compounds up to 1,000 times more potent have been identified,” Golde said.
On their mechanism of action, this past June Golde, Boris Schmidt of the Technical University of Darmstadt, Germany, and a number of other groups raised eyebrows when they showed that some γ-secretase modulators do not bind the enzyme, as most drugs do, but stuck to the substrate APP. They did so, no less, smack in the middle of APP’s Aβ sequence, at amino acids 28-36. At the Eibsee conference, Golde discussed follow-up data on this work. For example, the group’s finding implied that any compound reported to interact with this part of Aβ could, in theory, be a GSM. Indeed, many substances tested, even some peptides, proved to be just that. This includes, for example, the amyloid dye X-34 and, more potently, the pharmaceutical compound GSM-1.
Curiously, when the researchers mutated the Aβ28-36 site to confirm that GSMs really bound there, they also discovered that one particular mutation of amino acid 28 shifted APP cleavage so dramatically that Aβ42 practically disappeared from the mix of cleavage products. “If you had this mutation, you would not get AD,” Golde speculated. It might be interesting to see if such APP variants exist in cognitively sharp nonagenarians or centenarians.
In all, this study to date suggests three possible mechanisms of action for GSMs. They are shown to lower Aβ42 generation, they increase the relative amount of the short Aβ forms that interfere with aggregation, and they appear to inhibit aggregation directly by getting in the way of Aβ-Aβ binding. Which of these might be most important in an AD therapy is unknown, Golde said.
Importantly, GSMs of the future, as well as other therapeutic candidates that are wending their way through preclinical testing, are facing the conundrum that they shut off an upstream cause of AD but are being tested in humans at its downstream end, years after much of the damage has been done.
Golde joined a growing chorus of investigators voicing this concern. These researchers draw an analogy with heart disease, where statins prevent catastrophic outcomes but cannot mend a heart once it is failing after years of atherosclerosis and infarctions. That, in essence, is what the current trial design asks AD drugs to do, Golde said, even though scientists now generally believe that AD has a preclinical period that includes the development of its hallmark pathologies and extensive neuronal death before patients ever see a doctor. Along a similar vein, epidemiology suggests a preventive effect for NSAIDs and indeed for estrogen and statins, yet all failed in subsequent treatment trials. “There is a paradox where we must stop the trigger, but are testing preventive drugs in treatment trials,” Golde said.
Golde brought to this audience of mostly basic scientists a discussion from the clinical world of Alzheimer disease, which struggles to get out of a Catch-22. No proven disease-modifying drugs exist to justify the expense and risk of prevention trials, yet with FDA-approvable treatment trial designs, candidate drugs (which are likely best as preventives) can’t easily be proven to modify disease (see also ACT-AD workshop; ICAD trial design story). At present, Golde noted, the pharmaceutical industry is trying to work its way out of this bind in steps: develop a safe medicine, hope to show a statistically significant effect in treatment trials even if it is very small, then retest at the MCI stage and then retest for prevention, hoping for bigger effects along the way. This could take 25 years. Initiatives such as ADNI may speed up the process by forging consensus around biomarkers for use in diagnosis and trials. Likewise, DIAN, an international network for the study of autosomal-dominant FAD mutation carriers, is gearing up to characterize their presymptomatic decade with the same longitudinal battery of biomarkers. Golde called on the audience to become engaged in these issues. One solution, he concluded, lies in public-private partnerships to lower the hurdles for prevention trials, another in developing treatments for downstream targets, i.e., targets other than amyloid.—Gabrielle Strobel.
This is Part 2 of a seven-part series. See also Parts 1, 3, 4, 5, 6, 7.
Eibsee: Still Game for γ—Sparring With a Formidable Enzyme
At the 8th Eibsee Meeting on Cellular Mechanisms of Alzheimer’s Disease, held October 29 to November 1 in Germany, Bart de Strooper of the University of Leuven and Flaams Instituut voor Biotechnologie in Leuven, Belgium, set the scene for a session on γ-secretase, the unusual intramembraneous aspartyl protease that releases the Aβ peptide once the BACE enzyme is done clipping APP. γ-secretase, a complex composed of presenilin (PS), nicastrin (NCT), Aph1, and Pen2, remains a central target of drug discovery.
Before he got into new data, de Strooper looked back for a minute to say that the field’s extensive discussion of whether familial AD mutations in presenilin cause a loss or a gain of function in this enzyme in his view has been constructive, because it led to an important change of how researchers think about amyloid (ARF Live Discussion, follow-up discussion). “In understanding the toxicity of amyloid, we must consider not just its quantity, but the ratio of Aβ species,” de Strooper said. γ-secretase spews out a whole range of different Aβ species. How it does that is not clear; however, a model first proposed by Yasuo Ihara of Japan’s Tokyo University, whereby it starts cutting at a particular cleavage site and then perhaps cuts processively every three amino acids, is drawing some support in the field.
The main biological function of γ-secretase is Notch signaling, de Strooper said, and that is why inhibitors cause immunological and gastrointestinal side effects. “But I do not think the story of γ-secretase inhibition is over,” de Strooper said. γ-secretase is a highly complex activity, and drilling deeper may reveal more specific drug targets. De Strooper pointed out years ago that the different presenilin and APH1 genes that exist in the human (and also mouse) genome make up four different γ-secretase complexes (de Strooper, 2003). At the Eibsee, de Strooper presented data that distinguish among some of these, and made a case that Aph1b/c-bearing complexes might be targeted for AD drug discovery, whereas Aph1a-bearing complexes should be left alone to handle Notch cleavage. The widespread expression pattern of Aph1a compared to a more specific, brain-only expression of Aph1b/c in mouse, as well as Aph1b expression in human brain, is consistent with Aph1b playing a role in AD, de Strooper reported.
In a previous study looking for a physiological function of Aph1b/c, the group had deleted the gene. The knockout mice displayed deficits in prepulse inhibition, working memory, and a drug response to antipsychotics that was reminiscent of schizophrenia and, together, suggested that Aph1b/c-containing γ-secretase functions in neuregulin1 cleavage during brain development (Dejaegere et al., 2008). More recently, the group asked whether targeting Aph1b/c might rescue an AD phenotype in mice. To address the question, the scientists crossed their Aph1b/c knockout strain with Mathias Jucker’s APP/PS1 mice that develop aggressive early amyloidosis, and checked for amyloid load, memory and behavior, and γ-secretase inhibitor side effect profile. The crosses had far fewer plaques than did the APP/PS1 strain alone, and differed on all other readouts, as well. “This means these various γ-secretase complexes have slightly different biochemical activities, and suggests that some complexes may be better drug targets than others,” de Strooper said. By implication, a compound that blocks only Aph1b, not Aph1a, might change the Aβ species distribution without hitting Notch. However, whether Aph1b directly affects AD is unknown (see also Aph1b on Alzgene). These issues are being hotly pursued in several γ-secretase laboratories around the world.
The possible neurodevelopmental role of Aph1b/c-containing γ-secretase raises parallels with BACE. This secretase also has a dual role in cleaving APP as well as neuregulin during developmental myelination (see ICAD news story; Savonenko et al., 2008; see also Part 5 of this series).
Tough Love in the γ Club
The Eibsee meeting convenes a “hard core” of γ-secretase aficionados. One thing these labs do, in a friendly spirit of scientific verification, is subject one another’s newest data to independent replication—or lack thereof, as the case may be. Sometimes, it’s the Eibsee presentations themselves that get their share of needling at the same venue in later years. For example, the finding that a GxGD motif in presenilin is important for the activity of the enzyme complex (Haass and Steiner, 2002; Xia and Wolfe, 2003) has held up in the hands of other scientists (e.g., Sato et al., 2006). This year at the conference, Harald Steiner, in Haass’ group at the University of Munich, added to it by describing how his group probed the motif by replacing its conserved glycines at positions 382 and 384. This demonstrated that these glycines are crucial active site elements without which the enzyme can’t function (see, e.g., Fluhrer et al., 2008). Two other findings have a harder time withstanding collegial scrutiny. For example, Steiner also presented data showing that his group has been unable, thus far, to replicate a finding reported previously at the conference, namely that the protein tmp21 is a regulatory subunit of γ-secretase (see Eibsee story and Chen et al., 2006). Steiner noted that in his laboratory, several different purification regimens all suggest that small amounts of tmp21, and also the other proposed complex component cd147 (Zhou et al., 2005), may indeed loosely associate with the complex, but are unlikely to be true components of it.
The corrective element of independent reproduction emerged a second time when several groups noted that their own data failed to support a published role for the complex component nicastrin. Well received at the time, that work had proposed that nicastrin serves as a kind of gatekeeper that recognizes the substrate and then passes it on to the active site for cleavage (see Eibsee story). The idea was that substrate recognition requires a particular glutamate residue on nicastrin; however, follow-up experiments by de Strooper’s and Phil Wong’s groups cast doubt on this notion when it turned out that γ-secretase recognized its substrate just fine, thank you, even when this glutamate on nicastrin was mutated (Chavez-Gutierrez et al., 2008). At this Eibsee conference, Regina Fluhrer in Haass’ group at the University of Munich corroborated de Strooper/Wong’s finding by studying signal peptide peptidases, the simpler cousins of γ-secretase. These enzymes require no complex, but Fluhrer found that they, too, perform a kind of length measurement of the substrate’s extramembraneous stub that had been ascribed exclusively to nicastrin, suggesting substrate recognition could occur more directly by way of structural features between the substrate and the enzyme’s transmembrane domain. In essence, scientists at the meeting said that nicastrin is no longer seen as indispensable for substrate recognition.—Gabrielle Strobel.
This is Part 3 of a seven-part series. See also Parts 1, 2, 4, 5, 6, 7.
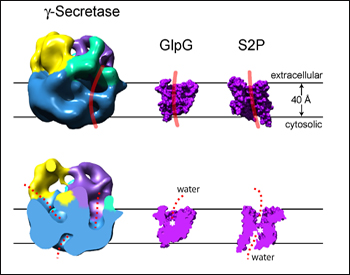
The mechanistic prodding and probing of γ-secretase would be much easier if only one could plainly see the thing. But to this day, the beast defies X-ray crystallographers trying to inspect its atomic structure. In the meantime, electron microscopists are edging ever closer to sneak better views. At the 8th Eibsee Meeting on Cellular Mechanisms of Alzheimer’s Disease held October 29 to November 1 in Germany, Huilin Li of Brookhaven National Laboratories on Long Island, New York, unveiled a second-generation EM image. It replaces a lower-resolution (15 Angstrom) EM picture derived with a negative staining technique with a new, slightly sharper one obtained with cryoelectron microscopy. CryoEM is considered superior for structural studies because it requires no contrast agents, hence affords a more “natural” view of the subject of interest.
The first structure (see Eibsee 2006 story), like the new one, came out of an ongoing collaboration Li has with Dennis Selkoe’s group at Brigham and Women’s Hospital in Boston. To move toward cryoEM, the researchers initially focused on obtaining a more concentrated and highly pure preparation of active human γ-secretase (Cacquevel et al., 2008). In the process of analyzing this preparation by scanning transmission EM, the collaborators, with co-first author Pam Osenkowski, confirmed recent work (Sato et al., 2007) demonstrating that γ-secretase occurs as a single complex containing each component once. The weight of each component plus that of the digitonin detergent added up to the 230 kD measured for the complex, Li reported at the Eibsee. Together, this appeared in the mind of the conference participants to settle previous questions of whether γ-secretase might contain two presenilins (Schroeter et al., 2003).
Li then noted that their next goal—seeing γ-secretase on cryoEM preparations—was technically challenging, consuming many months of effort. But eventually, co-first author Hua Li succeeded.
12-A resolution cryoEM structure of active human γ-secretase shown next to two known crystal structures of E. coli (GlpG) and archaeal (S2P) intramembrane proteases. Top: surface rendering of side view. The substrate might bind along the red line. Bottom: vertical view, cut open. Water might access the cavities in red. Image credit: Huilin Li
The new structure resolves the three-dimensional features of γ-secretase at 12 Angstroms. Globular and some 8.5 nm across, the enzyme at first glance looks simple and smooth on the cytosolic side and bumpier, busier on the extracellular side. It differs importantly from the first structure in that it no longer shows an inner chamber that reaches all the way through the membrane. This feature on the previous structure had inspired puzzled discussion of how pore- or proteasome-like features of γ-secretase could maintain the cell’s membrane potential. Instead of the hollow tunnel, the new cryoEM pictures show cavities that reach quite deep into the membrane from either side but do not connect. Overall, the new structure looks less like a channel and more like a protease, said Li.
The structure shows a groove across the membrane region that could be the proposed lateral gate through which the substrate slips on its way to the active site. It also shows various dents where water could enter to support the hydrolysis reactions the enzyme carries out within the hydrophobic environment of the membrane (Li et al., 2008).
Even so, this structure is still too blurry to settle topographic or mechanistic questions definitively, Li said. Next on his to-do list are cryoEM structures of γ-secretase cum substrate, and γ-secretase embedded in a lipid bilayer to give it a more native conformation. In the present preparation, the scientists cannot rule out that the high expression levels and the solvent might distort the complex into an artificial shape.
Other γ-secretase topics discussed at the conference included the in-vivo relevance of new cell-based findings by Jochen Walter’s group at the University of Bonn, Germany. Walter’s recent work using cultured wild-type, presenilin knockout, and transgenic cells suggests that γ-secretase might regulate cholesterol metabolism in neurons through LDL receptor-mediated uptake of lipoprotein particles (see Tamboli et al., 2008). Golde noted that one way to take such in-vitro findings into humans could be to measure their proposed consequences, for example, markers of cholesterol metabolism and ApoE levels, in people with autosomal-dominant FAD mutations (see DIAN study).—Gabrielle Strobel.
This is Part 4 of a seven-part series. See also Parts 1, 2, 3, 5, 6, 7.
Eibsee: Soft Cocktail—In Search of Gentle Knocks To BACE and γ
At the 8th annual Eibsee Meeting on Cellular Mechanisms of Alzheimer Disease, the theme of hitting two secretases at once to deal a blow to AD came up twice. Phil Wong of Johns Hopkins University, Baltimore, explored it with genetic experiments, and Stefan Lichtenthaler at the Ludwig-Maximilians University of Munich followed up with a small-molecule approach. For his part, Michael Willem, who is in Haass’s group at LMU Munich, added new data to the understanding of BACE’s physiological role, which is still in its infancy.
Wong got things going by arguing that while total inhibition of either APP-processing enzyme would lead to untoward side effects, moderately inhibiting both might be safe. Research by several groups has enlisted normal functions and substrates for BACE that make its wholesale block unsafe, Wong said. His own group used BACE knockout mice to show that this enzyme’s participation in neuregulin processing (Willem et al. 2006; Yan et al., 2006) partly explains why its complete loss leads to behaviors resembling certain endophenotypes of schizophrenia (Savonenko et al., 2008). Moreover, total BACE inhibition subtly impairs synaptic function, as well as BACE’s role in adult myelination of peripheral nerves (see ARF related news story). In contrast, paring back BACE by half reduced amyloid deposition while avoiding those side effects, Wong said.
On γ-secretase, the danger of inhibiting notch cleavage is well known. Wong’s group has used various different strains of heterozygous and homozygous knockout mice that ranged from 25 to 75 percent of normal γ-secretase activity to show that γ-secretase serves as a tumor suppressor in the skin and, at least in mice, can be safely inhibited globally by no more than a third. That was enough to reduce amyloid deposition (Li et al., 2007).
New at the Eibsee conference, Wong presented data on combining moderate inhibition of both BACE and γ-secretase. The idea behind this approach is that because both enzymes act on APP but the majority of their other substrates do not overlap, hitting both might enhance the desired effect and dilute the unwanted effects. To see whether this pans out, Wong’s group crossed mice heterozygous for both BACE and the γ-secretase component Aph1a with a mutant PS/APP strain and found that this moderate genetic reduction of both secretases achieved a greater attenuation of amyloidosis in the brain as compared to either of the single heterozygotes. The side effect profile was encouraging: myelination, a series of schizophrenia endophenotype tests used on prior knockout strains, were all normal, as was the skin and spleen, which are commonly affected in γ-secretase knockouts. These new mice are still aging but appear to have a normal lifespan. “ The therapeutic effect was additive, the side effects were not. This might be an attractive therapeutic strategy for AD, particularly for prevention, because these moderate reductions are not associated with toxic side effects,” Wong said.
Others at the conference agreed with this concept but pointed out that titrating the precise extent of the inhibition may be less practical in elderly humans than in genetic models. For example, some γ-secretase inhibitors have behaved paradoxically where low doses of inhibitor unexpectedly increased Aβ and only higher drug doses decreased the peptide (Lanz et al., 2006; Burton et al., 2008).
In his talk, Michael Willem of the University of Munich, noted that the potential risks of BACE inhibition as a drug target are only beginning to come to light. One controversy at this point is whether BACE participates in myelination both in the brain and peripheral nervous system, or only the latter. Willem’s ongoing research reinforces his and Christian Haass’s initial finding that BACE’s role in myelination plays out primarily in Schwann cells, not oligodendrocytes, hence is not needed for myelination in the brain (for a paper suggesting the opposite, see Hu et al., 2006). Recent crosses of BACE knockout mice to neuregulin heterozygote knockout mice showed profound hypomyelination, Willem reported, but again only of peripheral nerves.
But even if BACE1’s supporting role in myelination is predominantly peripheral, what that means for BACE inhibition in people with AD remains to be understood. Willem addressed this issue with LY-2434074, a BACE inhibitor made by Eli Lilly and company that will remain an experimental tool because the P glycoprotein transporter pumps it out of the brain too fast for it to be a serious drug candidate. When given to mice, this inhibitor effectively reduced Aβ levels in the brain and plasma of mice; however, the BACE substrate neuregulin accumulated along with APP, Willem noted. “Every substrate will accumulate to some extent if BACE is blocked,” he said. To what extent this will happen in the human brain is far from clear, in part because other proteases, including perhaps ADAM-17, appear to compensate partially for the loss of BACE1, Willem added (see also Sankaranarayanan et al., 2008). Neuregulin has many different functions in the nervous system. Many of them occur during development, but particularly BACE1s possible role in synaptic plasticity should be carefully studied in the course of pursuing BACE inhibition, Willem urged (see also Wang et al., 2008).
One-Two Punch: Can a Single Drug Block Two Enzymes?
Stefan Lichtenthaler picked up Wong’s concept of putting a mild damper on both β-secretase and γ-secretase in the hope of reducing amyloidosis safely. This would seem to require two drugs, as these two enzymes differ in all sorts of regards—where they are in the cell, how they cut, and what level pH they prefer, for example. But surprisingly, Lichtenthaler reported that he has come across compounds that he believes might eventually be able to pull off such a twofer. Lichtenthaler’s group ran a chemical genetics screen to look for small molecules that tweak APP shedding. Besides some known activators and inhibitors, the screen turned up bepridil, an old drug that is still being used in France and Japan to treat heart disease, particular chronic angina. This clinical record established that bepridil is safe for chronic use at plasma concentrations of some 3 to 10 micromolar. Bepridil has a role in blocking calcium channels and calmodulin. Curiously, closer examination of this drug showed that it is a dual modulator: it inhibits β-secretase and, separately, also tweaks γ-secretase, Lichtenthaler said.
The scientists first examined bepridil’s effect on BACE. In cell culture, bepridil blocks the enzyme with an IC50 of 6 micromolar. That is in the range achievable in human plasma in clinical practice, and bepridil is known to cross the blood-brain barrier. Whether this IC50 is potent enough to do much to BACE in the brain is unclear at this point; animal studies measuring Aβ levels in brain will address this question. (Again in cell culture, bepridil also blocked cleavage of neuregulin, Lichtenthaler noted.) To get a sense of how the drug might interfere with BACE1, the scientists first looked to its known role antagonizing calmodulin, but soon ruled that out. Instead, the group is pursuing a working hypothesis whereby bepridil affects BACE indirectly by raising the pH near the membrane of endosomes, where BACE is thought to work, upward from BACE’s low acidic comfort level.
Regarding bepridil’s effect on γ-secretase, the scientists found that it behaves like a typical “inverse” GSM. That is, in cells expressing the γ-secretase APP substrate C99, it boosted Aβ42, lowered Aβ38, and left unchanged Aβ40 and total Aβ. This, clearly, is the opposite of what AD scientists want from a γ-secretase modulator. Intriguingly, though, previous research by conference attendee Boris Schmidt of the Technical University of Darmstadt has shown that such inverse modulators can be flipped chemically to do the right thing. Turn a certain ester moiety into a carboxylic acid and, voila, you’ve got yourself an Aβ42-lowering GSM (Narlawar et al., 2007). Lichtenthaler is collaborating with Schmidt’s group to synthesize derivatives of bepridil that behave like a “straight” (i.e., Aβ42-lowering) GSM yet maintain their penchant for blocking BACE, as well. On his wish list of chemical characteristics for a perfect dual modulator are an aromatic core that lets the compound sink into the membrane to affect γ-secretase there, a COOH group to make sure the compound modulates the enzyme complex in the desired direction and, finally, an amine group necessary for upping the endosomal pH. At this stage, these studies are primarily geared to understanding the dual mechanism of bepridil and similar compounds. If this line research ever comes to pass as a dual-mechanism drug, one advantage would be that the patient would have to take one pill, not two, Lichtenthaler noted. —Gabrielle Strobel.
This is Part 5 of a seven-part series. See also Parts 1, 2, 3, 4, 6, 7.
Burton CR, Meredith JE, Barten DM, Goldstein ME, Krause CM, Kieras CJ, Sisk L, Iben LG, Polson C, Thompson MW, Lin XA, Corsa J, Fiedler T, Pierdomenico M, Cao Y, Roach AH, Cantone JL, Ford MJ, Drexler DM, Olson RE, Yang MG, Bergstrom CP, McElhone KE, Bronson JJ, Macor JE, Blat Y, Grafstrom RH, Stern AM, Seiffert DA, Zaczek R, Albright CF, Toyn JH.
The amyloid-beta rise and gamma-secretase inhibitor potency depend on the level of substrate expression.
J Biol Chem. 2008 Aug 22;283(34):22992-3003.
PubMed.
Eibsee: Antibody Binding Crystal Clear; New Vaccine in the Mix
At the 8th annual Eibsee Meeting on Cellular Mechanisms of Alzheimer’s Disease, held October 29 to November 1 in Germany, two speakers presented advances in the development of immunotherapies for AD. Despite widely reported hiccups in the clinic, this area of drug discovery remains among the most active in Alzheimer disease translational and preclinical research, and many scientists in the field share the hope that it can eventually succeed. At this conference, Guriqbal Basi of Elan Pharmaceuticals in South San Francisco, California, reported early findings of his group’s efforts to understand, with X-ray crystallography, why one particular antibody can have a therapeutic effect while another doesn’t. Ulrike Mueller of the University of Heidelberg, Germany, introduced a promising new go at crafting a highly immunogenic, safe vaccine.
Basi set up his new data by noting first that the original observation of AD immunotherapy—immunization with Aβ reduces amyloid pathology in mice—has been widely replicated. It has also been extended to other amyloid-related pathologies of AD in various model systems. Besides plaques, markers ranging from neuritic dystrophy, gliosis, vascular amyloid burden, to synaptic loss, deficits in long-term potentiation and cognition all can improve in response to the right antibodies, Basi said. At the same time, the question of precisely what constitutes the “right antibody” has come to the fore as one of the main mysteries to solve. Outwardly similar antibodies can behave quite differently in vivo, and the underlying characteristics of that are murky at best. Basi used comparative X-ray crystallography to drill down into why one antibody has a desired effect while another does not.
The scientists started from the observation that acute (aka passive) immunization with certain antibodies differs intriguingly from chronic (aka active) immunization with Aβ constructs, in that injected antibodies can have surprisingly rapid effects in both mice and slice cultures (Dodart et al., 2002; Shankar et al., 2007). As their behavioral assay to study a series of antibodies, Elan’s colleagues at Wyeth Pharmaceuticals chose contextual fear conditioning, where a mouse first learns an aversive stimulus, then remembers it. In this test, performance decline precedes age- associated increase in Aβ load. Interestingly, performance responds differently to otherwise similar therapeutic antibodies. Consider these three monoclonal antibodies by Elan, which all bind the Aβ3-7 N-terminal epitope that is widely believed to generate effective immune responses: the 12A11 potently reverses the fear conditioning deficit, the 10D5 antibody needs 30 times the dose of the 12A11 to get the same job done, and the 12B4 can’t do it at all (see also ADDF conference report).
What’s behind this difference? The binding affinities of these three antibodies to the same linear Aβ1-10 epitope did not correlate with their ability to reverse the cognitive deficit. What did correlate, however, was their ability to recognize oligomeric species of Aβ. In immunoprecipitation experiments, the most effective antibody pulled down much more Aβ dimers and trimers than monomer and larger aggregates, whereas the other two antibodies did not, Basi reported. The amino acid sequences of the three antibodies’ light and heavy chains differ only by some 5 to 15 percent.
Collaborating with Hadar Feinberg and Bill Weis at Stanford, the Elan scientists obtained crystal structures of the 12A11, the 12B4, the 10D5 and, for good measure, the 3D6 antibody that recognizes plaques and is the basis of the current series of Bapineuzumab (see Phase 3 immunotherapy trials). The scientists expressed these antibodies as recombinant Fab fragments and co-crystallized them with Aβ 1-7. Apologies, dear reader, the pictures and movies themselves have to await formal publication, but here is a brief impressionistic sketch in words: the structures show Aβ1-7 bound to antibody in an extended conformation in a groove atop the antibody. At 1.5 to 2.95 Angstroms, the structures’ resolution is high enough to allow the scientists to define the contact points between the antibody and Aβ residues that make up the binding. When overlaying the structures of the 12A11, the 10D5, and the 12B4 antibodies, low-resolution views betray little difference—the antibodies each appear to cradle Aβ in the same configuration. But zooming in to the contacts reveals that the minor amino acid differences in the antibodies’ sequences contribute to divergent contacts between antibody and antigen in the binding pocket. Most of these differences between the antibodies occur in the complementarity-determining region (CDR) 3 loop, Basi noted. Together, these atom-to-atom differences in the contacts between Aβ and antibody may eventually explain their different in-vivo properties, Basi said.
The group used X-ray structures to draw a different comparison, as well. This one was not between cognitively active and inactive antibodies, but between two antibodies that both recognize amyloid pathology in brain tissue but do so in subtly different ways. The 12A11 tends to bind the dense core of plaques, whereas the 3D6 tends to stain more diffuse plaque in frozen sections from PDAPP mouse brains, Basi said. Their Fab/Aβ structure looked very different from each other. Where the 12A11 captures the peptide in an extended conformation, the 3D6 holds a helical one. Taken together, crystal structures of four separate N-terminal Aβ antibodies illustrate differences in Aβ conformation that may correspond to its deposited forms, as well as subtle differences in antibody-antigen binding contacts that may give clues toward understanding cognitive effects of oligomeric forms of Aβ, Basi said.
These data largely fit with two prior reports of Fab-Aβ crystal structures of antibodies raised by independent investigators (Gardberg et al., 2007; Miles et al., 2008). These investigators also saw Aβ in an extended conformation and they also saw the same antibody CDRs contacting the peptide. A main difference is that these prior antibodies are not part of a therapy development program.
Many questions remain. How the CDR loop that houses most of the different contacts might relate to oligomer properties is unknown, as is whether the oligomer-binding antibody 12A11 can prevent neuronal toxicity or loss. A crystal structure of an antibody grasping an Aβ dimer is on the to-do list, as well, Basi said.
Also at the Eibsee meeting, Ulrike Mueller, now in Heidelberg, presented news from the other side of the immunotherapy spectrum. While Basi showed exquisite, near-atomic details of how a known monoclonal antibody interacts with Aβ, Mueller presented on a polyclonal immune response to a new active vaccine that was highly potent after a single shot.
Known for her sophisticated dissection of APP’s multiple functions in knockout and knockin mice (Ring et al., 2007), Mueller recently teamed up with card-carrying immunologists to join the fray with an AD vaccine of her own. Christian Buchholz at the Paul Ehrlich Institute in Langen, Germany, has for years worked to redesign retrovirus-like particles, from which all replication ability has been removed to render them harmless. Dubbed virus-like particles (VLP), they are derived from mouse leukemia virus (termed retroparticles). VLPs serve as display platforms that present densely stacked arrays of one’s antigen of choice to B cells and T cells (Buchholz et al., 2008). One of his papers, which reported that such an experimental vaccine could break the body’s tolerance to the prion protein (Nikles et al., 2005; Buchholz et al., 2006) caught Mueller’s attention, and the scientists, with Ulrich Kalinke also of the Paul Ehrlich Institute, subsequently collaborated to create a similar vaccine against Aβ. At the conference, Mueller shared results of first mouse studies with it.
The vaccine features retroparticles studded with several thousand copies of Aβ, Mueller said. When injected intravenously into 4-month old APP23 transgenic mice once, this vaccine by 11 months of age had elicited high levels of IgG1 and IgG 2b antibodies, the isotypes thought to avoid unwanted cytotoxic inflammatory reactions. Surprisingly, this single injection induced the same concentration of antibodies in the sera of the mice as did a series of six monthly booster shots. What’s more, the antibody response arose without use of an adjuvant. (Adjuvants have at times raised safety concerns when used in elderly people with multiple comorbidities.) Tolerance to human Aβ, which can keep antibody titers low, was not a problem with this vaccine. There were no signs of inflammation or cytotoxic T cell activation. The immune response in this study may have been effective and safe because VLP epitopes stimulate T cells directed against viral, not Aβ epitopes, Mueller said.
When the researchers analyzed the brains of the mice, they were surprised to see that the vaccine had reduced both plaque burden and the concentration of soluble pools of Aβ40 and 42. Other vaccines have been shown to reduce the former but, at least temporarily, raise the latter or leave it unchanged. “The sera from immunized mice bind to soluble Aβ on ELISA plates and to plaque Aβ on sections,” Mueller said.
Overall, Mueller said, her data on this new vaccine are consistent with those of the dendrimeric Aβ1-15 vaccine developed by Cynthia Lemere at Harvard Medical School, which is currently being studied in non-human primate models (see ARF related SfN story) and other, similar vaccines. The main differences at this early stage appear to be that Mueller’s vaccine appears to require no adjuvant and lowers soluble and fibrillar Aβ in parallel. The current study injected the retroparticle vaccine into mice before they had developed amyloid pathology, i.e., in a preventive mode. How it would perform in more advanced models that better reflect a typical human trial population is unknown at this point.
This latest candidate AD vaccine comes as part of a broader trend to develop VLP-based immunodrugs in medicine. This trend aims to deploy those particles against disease-related proteins for the treatment of chronic ailments (for reviews, see Jennings and Bachmann, 2008; Jennings and Bachmann 2008). In experimental settings, virus-like particles have been used to raise autoantibodies in models of arthritis, multiple sclerosis, and hypertension. Some are in clinical trials for HIV, Mueller said. Moreover, the approved prophylactic vaccine against cervical cancer is based on VLPs derived from human papilloma virus. The discovery of this virus’s role in cancer garnered half the Nobel Prize in Physiology or Medicine this past fall.
In Alzheimer research, virus-like particles already have left a small footprint. Four years ago, researchers led by David Morgan at the University of South Florida, showed that VLPs can help break tolerance against human Aβ in APP transgenic mice (Li et al., 2004) and Bryce Chackerian went on to show that VLP-based Aβ vaccines generate more desirable B and T cell responses than did some peptide-based vaccines such as Elan’s initial AN1792 (Chackerian et al., 2006, powID=56566.) What’s more, one VLP-based vaccine against Aβ, developed jointly by the Swiss biotechnology company Cytos and the pharma giant Novartis, was reported this past summer at the International Conference on Alzheimer’s Disease (ICAD) in Chicago to have appeared safe in initial human tests performed in Sweden. Called CAD106, this vaccine is currently recruiting for a one-year Phase 2 trial. (Notably, this trial is unusual in admitting people as young as 40 years of age—most AD clinical trials require participants to be at least 50 or 55, shutting out many patients with early onset AD.)—Gabrielle Strobel.
This is Part 6 of a seven-part series. See also Parts 1, 2, 3, 4, 5, 7.
Eibsee: A Step Toward Seeing Tau in the Living Brain
Last but not least, the 8th annual Eibsee conference, held this fall in Southern Bavaria, Germany, featured a poster session in which junior scientists presented new data. Topics ranged widely, from the molecular factors that control expression of the α-secretase enzyme (which scientists want to boost) to the use of D-enantiomeric peptides (which conveniently resist degradation by the stomach’s proteases) as a new therapeutic angle. To highlight but one example, a poster by Ali Taghavi struck into wide open—and coveted—territory. Taghavi, a Ph.D. student in Boris Schmidt’s medicinal chemistry laboratory at the Technical University of Darmstadt, proposed a chemical model that allows him to predict whether a given compound will more likely bind to Aβ or to tau pathology.
The purpose behind this chemistry research is to find a PET imaging agent for tauopathy. The goal of imaging tau pathology has become even more desirable since amyloid imaging in living people has reinvented biomarker research and handed the field a tool to better test the amyloid hypothesis in humans. The amyloid imaging field has flourished since the introduction of Pittsburgh compound B. A crop of competing tracers has sprung up, which are being used in rapidly growing clinical applications including ADNI and some clinical drug trials, and amyloid imaging has its own annual conference (see ARF HAI report; 2009 announcement). The coexistence of Aβ and tau characterizes AD pathologically; hence, PET imaging of Aβ alone is insufficient for a diagnosis of AD. In addition, tau pathology occurs in a range of other dementing diseases for which a live imaging tool could be hugely valuable. But in stark contrast to amyloid imaging, tau imaging is not out of the gates yet. It is not for lack of trying. Many labs are working on the problem, but a sufficiently selective chemical with the right characteristics of binding and brain penetrance has remained elusive to date.
Taghavi in 2006 received a graduate student stipend by the Breuer Foundation that also supports the Eibsee conference. He set out to understand what makes ligands selective for either tau or Aβ. For a benchmark, he started out with IMPYs, thioflavin T-like compounds for whom the relationship between their structure and their activity in binding Aβ fibrils was published (Zhuang et al., 2003; Cai et al., 2007). The published data suggested that the ligand inserts between the tracks of the Aβ fibril in such a way that the aromatic part of the ligand interacts with side chains of the Aβ fibril. Importantly, such a binding mode would not tolerate bulky groups sticking out, but it would tolerate both amphiphilic and non-amphiphilic compounds. (Amphiphilic means that the ligand has one polar, i.e., hydrophilic, and one non-polar, i.e., hydrophobic end, as lipids do, e.g.) In other words, Aβ fibrils would not “care” whether a ligand is amphiphilic or not so long as the ligand was slender and flat enough to align with the narrow channel that forms the binding site on the fibril.
What about tau? Taghavi’s first round of studying the structure-activity relationship between prospective ligands and tau showed right away that amphiphilic ligands bound, whereas non-amphiphilic ones did not. Subsequent fluorescence experiments, too, suggested that an amphiphilic character is necessary for a prospective ligand to bind to paired helical filaments of tau (PHFs are the structural constituents of tangles). In addition, PHFs seemed to prefer ligands of a different shape than did amyloid fibrils. For one, bulky groups on the hydrophilic end were not a hindrance to PHF binding. There was, however, a steric (or spatial) limitation to the hydrophobic end of the ligand. This hinted that, in binding tau PHFs, an amphiphilic ligand inserts into a pocket of a limited size with one end, and sticks out of the PHF with the other. In essence, tau ligands can be imagined almost like the letter T, or like a pin going into a pincushion, where the sharp hydrophobic pin enters the PHF while the hydrophilic button sticks out, facing an open space. Inside the PHF, the pocket for the pin is tight and rigid, holding only straight and rigid ligands, whereas the hydrophilic space can accommodate big polar groups. “To my knowledge, this is the first proposed model for tau-PHF activity of a prospective ligand,” Taghavi said in an interview.
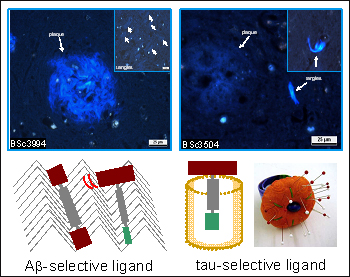
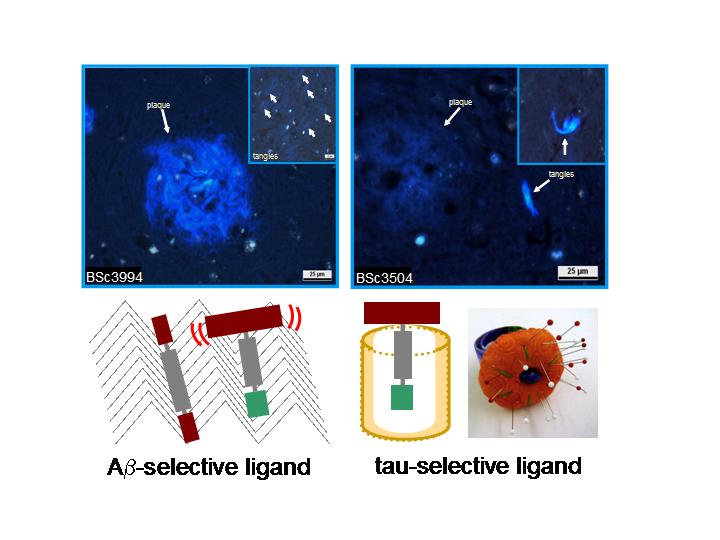
Left: Aβ-selectivity was achieved with non-amphiphilic flat and rigid ligands. Staining with BSc3994 resulted in strong staining of large blue fibrillar plaques (in the center) and weakly stained tangles (small box). Right: Tau-selectivity was achieved with T-shaped amphiphilic ligands, whose bulky polar group prevents plaque binding. BSc3504 exhibited strong tangle staining and left plaques unstained. View larger image. Image credit: Ali Taghavi
To test his model, Taghavi made a second generation of tau PHF ligands. He varied side chains of IMPY-based ligands to get a sense of just how bulky the polar end can get while retaining strong tau binding. In the process, he synthesized compounds that bind with an IC50 of 2 to 5 micromolar in two different tau assays done in collaboration with Eva-Maria and Eckhard Mandelkow at the Max-Planck Unit for Structural Molecular Biology in Hamburg. Next, Taghavi set aside IMPYs and expanded the chemical synthesis to other compound classes. One such ligand, called BSc3504 (read as “synthesized in Boris Schmidt’s lab”), binds with an IC50 just below the micromolar range. Its bulky polar end precludes binding to Aβ fibrils, and it lights up tangles brightly on tissue sections from AD brain. Further compounds are in the works.
At this point, the technical gap that needs bridging is that Taghavi does not yet have quite the right tau affinity assay. “It is not clear which form of tau you want to show affinity against. It’s not clear what the target should be. I used three tau targets: tau aggregation, PHF depolymerization, and human tissue fluorescence. I believe our best compounds are good PET ligands,” Taghavi said.
Incidentally, the model for selectivity might help explain why some amyloid imaging agents are more selective for Aβ than others. For example, PIB is non-amphiphilic; hence, the model would predict it does not bind tau (it doesn’t), whereas FDDNP has a polar group on one end that makes it amphiphilic and predicts binding of both Aβ and tau (it does; see Braskie et al., 2008). In the last three years alone, a wealth of amyloid imaging agents have sprung up, creating intense scientific and commercial competition (for a recent review, see Klunk and Mathis, 2008). Imaging of tau, or α-synuclein or TDP-43, for that matter, remains entirely up for grabs.—Gabrielle Strobel.
This is Part 7 of a seven-part series. See also Parts 1, 2, 3, 4, 5, 6.
{kind=link}
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.