The 67th American Academy of Neurology conference drew more than 12,000 attendees to Washington, D.C., April 18 to 25. Among the sessions for practicing neurologists, some research snippets stood out, including imaging biomarkers for tracking the progression of Huntington’s and Parkinson’s diseases and the identification of proteins in the skin that are normally associated with brain pathology.
Davies, Sperling Share 2015 Potamkin Prize
Two researchers share the 2015 Potamkin Prize for Research in Pick’s, Alzheimer’s, and Related Diseases. Peter Davies from the Feinstein Institute for Medical Research in Manhasset, New York, and Reisa Sperling of Brigham and Women’s Hospital and Massachusetts General Hospital in Boston received the award at the 67th annual meeting of the American Academy of Neurology, held in Washington, D.C., April 18-25. They will split the $100,000 prize, which comes from the Potamkin Family Foundation of Miami Lakes, Florida, and rewards contributions to research in neurodegenerative disease.
Honored:
Peter Davies, left, and Reisa Sperling share this year’s Potamkin Prize for Research. [Images courtesy of the Feinstein Institute and MGH.]
Davies was instrumental in first describing the loss of cholinergic neurons in Alzheimer’s disease (see Davies and Maloney, 1976). That work led to the development of anticholinesterase drugs, one of only two types of treatment the FDA has approved for Alzheimer's. Davies later focused on tau, developing a host of tau antibodies including the well-known Alz50 and MC-1, which remain among the most widely used in the field. Davies developed mouse models of tauopathy and championed the idea that this microtubule-binding protein can reactivate the cell cycle in neurons in people with AD (Andorfer et al., 2005). Most recently, Davies has focused on post-translational modifications and conformational changes in tau that could contribute to disease, as well as tau therapies (see May 2011 news).
Sperling has focused her research on early detection and treatment of AD. She is the principal investigator of the Harvard Aging Brain Study and previously led the joint effort by the National Institute on Aging and Alzheimer’s Association to propose new guidelines diagnosing preclinical AD (see Apr 2011 news on Sperling et al., 2011). She leads the A4 Study, a three-year secondary prevention trial that is testing the anti-Aβ antibody solanezumab in cognitively normal older people who have amyloid in the brain (see Dec 2014 news).
At AAN, Sperling gave an update on A4, saying that 1,400 people have been screened and more than 100 randomized thus far. She announced that an A5 trial will be underway soon. It will test Janssen’s BACE1 inhibitor in people 60 and older with amyloid accumulation. Both Davies and Sperling have been scientific advisers and frequent contributors to Alzforum. Congratulations!—Gwyneth Dickey Zakaib
New PET Tracer Tracks Preclinical Movement Disorders
After years of searching for diagnostic biomarkers for neurodegenerative diseases, scientists more recently have begun focusing on markers that change as these disorders progress. Such markers could indicate if a treatment works. Two imaging markers that might track Huntington’s and Parkinson’s diseases even before symptoms set in stood out at the 2015 annual meeting of the American Academy of Neurology, held this year from April 18 to 25 in Washington, D.C. Both tracers bind predominantly in the striatum. The evidence is preliminary, but experts agreed it looks promising, especially for monitoring the earliest stages of both HD and PD.
Huntington’s disease takes a particular toll on the medium spiny neurons (MSNs) of the striatum. These neurons produce gobs of phosphodiesterase 10A, an enzyme involved in cell signaling. Because other neurons in the brain express PDE10A at lower levels and because it is one of the first proteins to wane in mouse models of HD, PDE10A makes an attractive biomarker candidate (see Hebb et al., 2004). The mutant huntingtin protein may also interact directly with the transcription machinery for PDE10A to reduce its expression (Hu et al., 2004). “The thought has been if we can track PDE10A expression over time, we will get a picture of MSN degeneration in the striatum,” said Sarah Janicki, Columbia University Medical Center, New York. “That may give a unique picture of brain changes in Huntington’s.”
At Molecular NeuroImaging (MNI) in New Haven, Connecticut, a privately held neuroimaging services company, a research group led by David Russell has developed an F18-labelled PET tracer for PDE10A called MNI-659. Data from humans and non-human primates suggests that the tracer specifically binds in brain areas known to express PDE10A, and in vitro assays reveal that MNI-659 competes for known inhibitors of PDE10A activity, Russell told Alzforum (see also Barret et al., 2014).
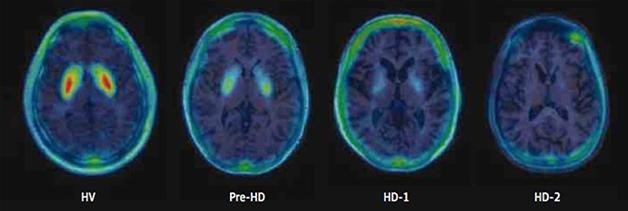
In a previous cross-sectional study, Russell found that in 11 carriers of the HD gene, loss of MNI-659 signal correlated both with the Unified Huntington’s Disease Rating Scale scores and the burden of pathology score. The latter score can estimate disease burden in mutant huntingtin carriers based on their CAG repeat length and age (see Russell et al., 2014). The MNI-659 signal dropped minimally in asymptomatic carriers, more in mildly symptomatic stage 1 patients, and still more in patients in stage 2 (see image below). This sample did not include people with moderate- to late-stage HD, i.e. stages 3 through 5. Two of the three people without symptoms registered at the low end of normal signal, while another had lost a bit of signal, suggesting that the tracer picks up change before clinical symptoms appear. “This might be one of the first biomarker indications of changes to come in HD,” Russell told Alzforum, perhaps even preceding striatal degeneration.
At AAN, Russell presented early longitudinal results on eight of those people. Two asymptomatic volunteers were five to 10 years from onset; three more people had stage 1 disease, and three stage 2. For all, the signal in the putamen and the caudate nucleus—the major divisions of the striatum—dropped over one year, regardless of the stage of disease. In the caudate, subjects averaged a 16.59 percent reduction per year, compared to 6.9 percent in the putamen and 5.81 percent in the globus pallidus. The rate of decline appears to continue, as the researchers have begun scanning these people for a second year of follow-up. So far, data on four participants suggest binding declines again by about 5 to 6 percent per year in the putamen and globus pallidus, and 15 to 20 percent per year in the caudate. At the earliest time points, lowered PDE10A expression likely accounts for most of the loss of signal, Russell said, while later on, dying neurons probably contribute.
To compare this rate of loss with healthy controls, Russell has taken single PET scans of a cohort of healthy controls. This cross-sectional data hints at an age-dependent loss of signal amounting to less than 1 percent per year in all striatal regions. That means the loss for HD patients speeds up at least five- to 15-fold, he said.
Given that MNI-659 binding declines early and differences are measurable within just a year, Russell believes it could make a good biomarker for striatal pathology in HD. It seems to start dropping in pre-symptomatic to early disease stages, meaning it may pick up on a crucial time window in HD, he added. The agent may be best suited for early disease, he told the audience, as binding appears to bottom out toward middle stages. One patient with stage 2 disease registered almost no signal, he pointed out.
MNI-659 could complement volumetric magnetic resonance imaging (MRI) and PET with fluorodeoxyglucose, which may be more informative later on, said Russell. A larger, longer, multi-site trial of MNI-659 with more patients representing a wider range of disease stages is needed.
Russell will test how this marker changes in Parkinson’s disease, or in other disorders that attack the striatum, such as schizophrenia or addiction.
Audience members asked know how MNI-659 PET related to MRI and FDG-PET. Russell said that he had no longitudinal MRI data, but previous cross-sectional study suggested that striatal volume decreased essentially in parallel with MNI-569 signal loss. However, MNI-659 seems to change first, as people with HD who still had normal striatal volume already registered MNI-659 loss. As for FDG-PET, Russell has no data on that marker to compare with MNI-659, but pointed out that it is known to mirror volume declines (see Tang et al., 2013). In Alzheimer’s disease and also frontotemporal dementia, the use of FDG-PET is declining as more data are becoming available for more disease-specific tracers of amyloid and tau pathology, respectively.
“These data looked really strong,” said session co-chair Victor Sung, University of Alabama School of Medicine, Birmingham. He asked how PDE10A inhibitors, which are being developed as a potential treatment for HD (Fusco and Giampà, 2015), would affect MNI-659 imaging. Russell said that since the drugs bind to the same region of PDE10A, they would likely displace the tracer.
Janicki agreed that the data were encouraging. “MNI-659 appears to be both sensitive and specific for the striatum,” she told Alzforum. She wants to know when signal starts to drop in pre-manifest patients relative to disease onset. “Hopefully this could be used to identify people most likely to convert [to symptomatic disease], so we might intervene as early as possible,” she said.
Parkinson’s and Beyond
Researchers at MNI are also hunting for imaging markers for PD progression. John Seibyl presented two-year follow-up data from 241 newly diagnosed patients in the Parkinson’s Progression Markers Initiative (PPMI) who underwent dopamine transporter (DaT) SPECT scans with 123-I-Ioflupane. This older tracer measures levels of the dopamine transporter. Researchers focus on signal loss in the striatum, as they do for MNI-659. However, whereas MNI-659 binds PDE10A produced in postsynaptic terminals of the striatum, the dopamine transporter appears on presynaptic terminals of axons that originate in the substantia nigra. A deteriorating 123-I-Ioflupane signal indicates a loss of those dopamine neurons.
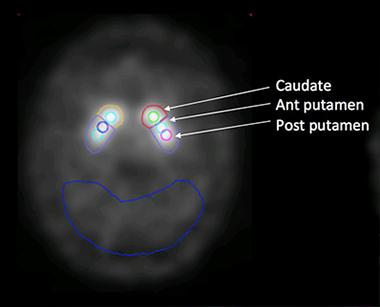
Seibyl and colleagues examined change in the striatum as a whole, and in its component regions, i.e. the caudate, anterior putamen, and posterior putamen. They compared brain regions contralateral of the side of the body most affected by PD symptoms with brain regions on the ipsilateral side, which would be presumed to have less pathology (see image below).
Regions of Interest:
The researchers look at tracer uptake in both sides of the striatum. The cerebellum (outlined in blue) serves as the reference.
[Image courtesy of John Seibyl.]
In all patients, the contralateral (i.e., the more affected) side took up less tracer at baseline than the ipsilateral side. A year later, uptake in the contralateral side had fallen another 7.4 percent, and 15 percent by year two. Though the less affected side retained more tracer at baseline, it too lost binding capacity—11.8 percent in the first year, and 17.1 percent total over two years. This suggests that in newly diagnosed patients, the striatum connected to the less-affected half of the body loses relatively more DaT binding than the contralateral side. The biggest decrease seems to happen early in the disease, slowing down later on.
Analyzing each striatal region individually, the researchers found that those on the ipsilateral side followed a consistent pattern of signal loss: The more posterior the region—the posterior putamen, followed by the anterior putamen, and then the caudate—the greater the loss. The pattern on the contralateral side was less clear. There, the posterior putamen lost the least signal, compared with the other striatal regions. Seibyl said he is unsure why that would be.
How does the reduction in binding correlate with disease? Change in DaT scan signal did not correlate with decline on the Unified Parkinson’s Disease Rating Scale. This is not unusual, Seibyl told Alzforum, as biomarkers and clinical motor ratings measure different aspects of neural function.
The results hint that DaT scans could be a useful marker of early PD progression, but more work is required to figure out what a changing signal means, Seibyl said. Already, it appears as though researchers should consider striatal regions separately. “If you were designing a clinical trial of a putative neuroprotective compound, where in the striatum you sample may be important for determining the effect,” Seibyl said. Russell agreed. “The most prognostic information at baseline may be in the contralateral putamen, but to follow disease progression from there, the caudate or the ipsilateral putamen may be better since they won't ‘bottom out.’”
Just as in HD and PD, data are trickling in on molecular imaging in AD, particularly tau pathology. Brad Dickerson, Massachusetts General Hospital, Boston, presented data on the PET imaging ligand T807, much of which Alzforum covered at the Human Amyloid Imaging conference earlier this year (see Feb 2015 news). Dickerson presented one-year follow-up data on a person with frontotemporal dementia. He reported a tight correlation between the T807 binding pattern at baseline and at 12 months, suggesting good test-retest reliability. Binding increased by as much as 10 percent in some areas, hinting that the scan can detect accumulation over time. In addition, some areas that were bare at baseline but lay near affected regions—such as the medial temporal gyrus and supramarginal gyrus— took up tracer one year later, suggesting that the pathology had spread. From the perspective of tracking treatment, that is encouraging, suggested Dickerson. “If we had a disease-modifying therapy, we might be able to show efficacy if we detect reduction in that increase over time,” he said.—Gwyneth Dickey Zakaib
Neurodegenerative Proteins in the Skin Could be Diagnostic
Neurodegenerative disease proteins hide behind the protective barriers of the central nervous system, complicating attempts to detect and track disease. While brain imaging and cerebrospinal fluid analyses detect some of these proteins, the techniques are complex, invasive, or expensive. Blood, though easy to obtain, has proven a poor source for neurodegenerative disease markers. What about the skin? At the annual meeting of the American Academy of Neurology, held April 18 to 25 in Washington, D.C., researchers reported that protein aggregates found in the brains of people with amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and Alzheimer’s disease (AD) appeared in the dermal layers, too. More research is needed to figure out what this could mean, but some think the skin could become a source of diagnostic markers.
In the developing embryo, brain and skin cells arise from the same ectodermal germ layer. Some scientists hypothesize, therefore, that some protein abnormalities found in neurons may also arise in the skin. In PD patients, deposits of α-synuclein were found in the nerves of the skin, though not in skin cells themselves (Doppler et al., 2014; Donadio et al., 2014; Wang et al., 2013). Other studies reported that skin changes can precede neurological problems in PD and ALS. For instance, people with PD had a greater incidence of melanoma, while some people with ALS were reported to have leathery, less-elastic skin that seemed to resist bedsores (Matsuo and Kamitani, 2010; Ono, 2000). It remains unclear if these conditions are related to the toxic proteins that accumulate in the brain, but some researchers are studying the question.
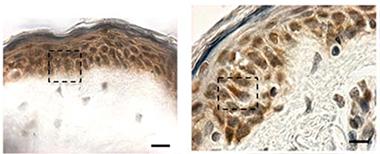
Hiding in Plain Sight:
Cytoplasmic TDP-43 inclusions (reddish brown) are absent in biopsied skin from healthy controls (left), but show up in patients with sporadic ALS (right). [Image courtesy of Paré et al., 2015.]
At AAN, Nicolas Dupré of Laval University, Québec, presented data from findings he and colleagues published last January. According to their study, TDP-43 forms cytoplasmic inclusions in fibroblasts from ALS patients (Paré et al., 2015). Senior author François Gros-Louis took arm skin biopsies from six people in each of three groups—healthy controls, people with sporadic ALS, and people who carried a hexanucleotide expansion in the C9ORF72 gene that causes ALS and frontotemporal dementia (five of whom were presymptomatic). They found TDP-43 aggregates in skin cells from all but the healthy controls (see image at left). Cytoplasmic TDP-43 inclusions are considered a hallmark of pathology; the protein normally resides in the nucleus (see Jan 2010 news story). Previously, researchers had reported elevated TDP-43 in the nuclei of skin cells from ALS patients (Suzuki et al., 2010).
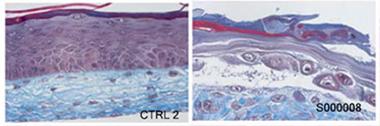
Gros-Louis and colleagues did not stop there. They work closely with a lab that grows human-derived skin for burn victims, and hence decided to engineer the same kind of tissue from the biopsied material and see how it differed from normal skin. They isolated skin fibroblasts and grew them in individual layers. Then, placing one atop the other, they let them fuse. They added a layer of keratinocytes to form an epidermis. Two weeks later, the researchers had a three-dimensional model of human skin. Derived from healthy controls, the new skin developed a fully differentiated epidermis and neatly organized dermis. On the other hand, tissue grown from either C9ORF72 mutation carriers or sporadic ALS patients formed an undifferentiated epidermis with poorly fused layers (see image below). The structural skin protein collagen appeared disorganized, as well.
Model skin:
Tissue-engineered skin from healthy controls (left) has a well-developed dermis (blue) and epidermis (purple), unlike that from a C9ORF72 patient (right). [Image courtesy Paré et al., 2015.]
The TDP-43 antibody 12892-1-AP revealed the cytoplasmic aggregates of ALS pathology in almost a third of the cells of the synthetic skin derived from each patient. The cytoplasmic inclusions appeared both in tissue grown from symptomatic sporadic patients, and from pre-symptomatic C9ORF72 mutation carriers. By contrast, only 4 percent of cells in engineered skin from healthy donors had them. That the abnormalities cropped up in skin made from pre-symptomatic patients suggests that they may predict oncoming neurological disease. However, the researchers are unsure when these proteins accumulate in skin relative to the onset of neurological symptoms.
Because it can be cultured ad infinitum, the tissue-engineered skin could eventually offer a renewable source of human tissue to use in the search for reliable biomarkers, to test diagnostic methods, and perhaps to monitor the effectiveness of potential treatments, speculated Dupré, though more research is required.
Pathogenic proteins may appear in skin from AD and PD patients, as well, according to research led by Ildefonso Rodríguez-Leyva, Central Hospital, Ignacio Morones Prieto, San Luis Potosi, Mexico, and senior author María Jiménez-Capdeville, Universidad Autónoma de San Luis Potosi. At AAN, they reported recently published findings that α-synuclein and phosphorylated tau (p-tau) aggregate in the skin of people with PD, while p-tau also turns up in the skin of AD patients (Rodríguez-Leyva et al., 2015; Rodríguez-Leyva et al., 2014).
The researchers took small skin biopsies from behind the ears of 12 healthy controls, 16 people with PD, 17 with non-neurodegenerative dementias, and 20 with AD (diagnosed using NINCDS-ADRDA criteria; McKhann et al.,1984). The researchers tested the tissue with the antibodies AT8, which detects tau phosphorylated at serine 202 and threonine 205, and E178, which recognizes tau phosphorylated at serine 396, as well as non-phosphorylated tau. They also used RB-9026-P, a polyclonal antibody for α-synuclein. Immunohistochemistry and western blots indicated that cells from AD and PD patients bound seven and five times more AT8, respectively, than control cells. E178 detected no differences among the tissues, suggesting that total tau levels were unchanged. PD skin cells also had up to seven times more α-synuclein than control tissue.
Other scientists had previously observed phosphorylated tau in the brains of PD patients (Wills et al., 2010).
“These are preliminary findings and will have to be reproduced in larger groups of subjects with and without the diseases of interest,” said John Hart, University of Texas at Dallas, who co-chaired the symposium.
Jiménez-Capdeville will next check whether Aβ is present in patients’ skin cells, and whether the amount of any of these proteins increases over time in parallel with pathogenic processes in the brain. As with TDP-43, the scientists do not know when these proteins turn up in the skin relative to the onset of disease.
Skin cells could potentially offer a ready source of biomarkers for neurodegenerative diseases, said Tapan Khan, Blanchette Rockefeller Neurosciences Institute, Morgantown, West Virginia. Skin cells can be cultured and resampled. Khan is working on skin biomarkers for Alzheimer’s disease. He thinks cell signaling changes that precede protein aggregation in skin fibroblasts could catch disease earlier than waiting for those proteins to accumulate. His work suggests reduction in protein kinase Cε could herald AD in these cells (see Khan et al., 2015).
“The findings are interesting, but the authors have a lot more work to do to move these approaches further,” said John Trojanowski, University of Pennsylvania, Philadelphia. For starters, the authors need to confirm via biochemistry that the antibodies are truly picking up the neurodegenerative proteins in question. “The skin is notoriously fraught with artifacts because of non-specific binding of antibodies,” Trojanowski said. The sample sizes in these studies need to become larger, and subjects need autopsy confirmation of their underlying diseases, he said.—Gwyneth Dickey Zakaib

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.