Past Webinar
Generation of an Apoptotic Intracellular Peptide by γ-secretase Cleavage of Alzheimer's β-amyloid Precursor Protein
Quick Links
Introduction
Benjamin Wolozin, with Luciano D'Adamio and Eddie Koo, led this live discussion on 20 September 2000. Readers are invited to submit additional comments by using our Comments form at the bottom of the page.
Journal of Alzheimer's Disease. 2000; 2(3-4): 289-301.
Brent Passer1*, Luca Pellegrini1*, Claudio Russo2, Richard M. Siegel3, Michael J. Lenardo3, Gennaro Schettini2, Martin Bachmann4, Massimo Tabaton5 & Luciano D'Adamio1,6
Transcript:
Live discussion held on 20 September 2000 and moderated by Benjamin Wolozin.
Participants: Benjamin Wolozin, Luciano D'Adamio, Ed Koo, Mark Smith, Donna McPhie, Soshi, Guest 3, June Kinoshita.
Note: Transcript has been edited for clarity and accuracy.
BWolozin: Okay, I believe that I am moderating this session, so let's Begin.
Eddie Koo: Good morning to everyone. It's still kind of early here.
BWolozin: Luciano, perhaps you could begin by stating your hypothesis.
Luciano: OK Ben. We are studying the role of the three FAD genes in the regulation of programmed cell death. Initially, we isolated a dominant negative form of ALG3 as an inhibitor of Fas-induced apoptosis. ALG3 is lso an inhibitor of the gamma secretase activity mediated by presenilins. The model we propose in this manuscript is that presenillins regulate the sensitivity of cells to apoptosis by controlling the release of the intracellular APP domain, that we have named AID. This biological role of presenilins and APP might underlie the pathogenic mechanisms of AD. In fact, conditions that cause increased APP processing cause AD but also sensitize cells to apoptosis by increasing the amount of AID that is produced.
MSmith: Hello everyone...is Perry on line?
June Kinoshita: No. He's in Italy... must be off having a bottle of Chianti. Just for clarification, is AID a precursor of C31, or is it the same thing called by two names?
Luciano: AID is the intracellular (c-terminal) APP fragment generated by gamma secretase (59 AA long). C31 represents the last 31 amino acids of AID.
Eddie Koo: Any APP C-terminal fragment is a potential precursor of C31. We never tested AID as the precursor, but certainly C99/C100 and C83 are precursors. No reason why AID cannot be but detecting the fragment of AID cleaved by caspase would be exceedingly difficult.
Eddie Koo: Luciano's hypothesis is consistent to what's been proposed by others. We know neurons die in AD but how and why is the $64[K] question.
June Kinoshita: There appears to be a growing family of APP-derived fragments that have toxic effects in vitro.
Eddie Koo: Luciano's results are quite interesting. Between the original results of Bruce Yankner and Rachael Neve, followed by all the other C99/C100 toxicity, our caspase results, and Luciano's AID all suggest that in vitro at least, something funky is going on in the C-terminus.
June Kinoshita: That is, there's more to it than just Aβ?
Eddie Koo: Well, June, that's also part of the equation. Is it just Aβ or more than Aβ. Although Luciano's results are different from ours, neither of our results implicates Aβ directly.
Luciano: Our studies indicate that the processing of APP, and in particular, the fragments released by gamma secretase have the biological role of modulating the threshold of cells to cell death.
Luciano: I agree with Eddie's remarks. Our results indicate that the last 31 C-terminal amino acids of APP are inert with regards to apoptosis. However, something is going with this C-terminal region of APP. The important concept is that gamma secretase can regulate programmed cell death.
MSmith: Luciano.....In vitro or in vivo?
Luciano: Mark, I think in vivo.
June Kinoshita: So this C31 (if I may call it that) fragment doesn't directly interact with an apoptotic pathway, but potentiates it?
Eddie Koo: Our original interpretation of the results is that C31, when formed, potentiates cell death. Luciano's experiments are the first to attempt at repeating our experiments and unfortunately is negative. But the conditions are not the same as ours.
Luciano: I agree with Eddie that the systems are different. We can't exclude that C31 has proapoptotic activity. In our hands it doesn't, while AID is highly toxic. We think that processing of AID by caspases represents a negative feedback mechanism that inhibits the toxicity of AID.
Eddie Koo: I don't understand Luciano's comment about negative feedback. If that's the case, then the AID mutant that cannnot be cleaved by caspase should be altered, which I believe is not.
Luciano: Eddie, the AID mutants (that can't be cleaved by caspases) are, in our hands, always more toxic. We are now attempting to quantitate this difference by using systems other than transient transfection.
June Kinoshita: Is there any evidence to support an in vivo role for C31?
Eddie Koo: We can only see the hallmark of APP caspase cleavage in AD brains.
Soshi: Do any of these toxic peptides occur in vivo at levels that can cause apoptosis? All in vitro experiments [involve] overexpression.
Eddie Koo: I agree with Soshi. The potential achilles heel of all the experiments is the overexpression. Where this fits in physiological levels is totally unknown.
Luciano: Soshi, this is an important question. The problem is that AID has a half-life of only ten minutes, so it is very difficult to quantitate. Besides the experimental differences, we agree with Eddie that the C-terminal APP region has important signalling properties that regulate apoptosis.
Eddie Koo: The one argument against AID in vivo is that in the PS knock in animals, Bob Siman cannot see any cell death. Thus, when more gamma 42 cleavage occurs, it doesn't seem to matter, at least to mouse neurons.
June Kinoshita: What differences might be critical between Eddie's experiments and Luciano's?
Soshi: I think the only toxic peptide known at high levels in brain is β amyloid. What does Dr. Koo think?
MSmith: Is amyloid toxic?
June Kinoshita: There you go again, Mark!
June Kinoshita smiles
Eddie Koo: I'm not really in the Aβ toxicity camp. I cannot believe for that amount of amyloid load in brain, the "toxicity" is really not that substantial in my mind.
Luciano: Yes, Eddie. I think the FAD mutations alter AID production to a measure that is not sufficient to directly kill neurons (otherwise these mutations would not be compatible with life). They might just make neurons more sensitive to apoptotic stimuli, i.e. you might need to trigger apoptosis to see any differences.
Eddie Koo: The key issue is that neurons are dying somehow, regardless of the artificial necrosis or apoptosis distinction. I think Aβ toxicity, if true, is only part of the answer.
Eddie Koo: Back to Bob Siman's animals: he reported that he was unable to kill the neurons with the insults he used. I cannot recall but probably excitotoxicity, ischemia, etc. But the neurons did not appear sensitized. Although Mark Mattson did report some sensitivity in his knock in animals, the changes were subtle.
Luciano: Our data do not exclude a role for Aβ;. They suggest that another APP-derived peptide (AID), which is stoichiometrically produced along with Aβ, is toxic and might participate in the neurotoxicity in AD.
Eddie Koo: I would say Luciano's statement is a good summary and a very reasonable interpretation. The $64K again is which of the several in vitro models are physiologically relevant. And this, I don't think anyone can say yet.
MSmith: Sorry guys..this computer is driving me crazy....would love to stay and discuss the nuances of amyloid toxicity (sic) and rampant apoptotic cell death in AD (double sic!) but cannot see what you are all saying...toodle pip!
Luciano:Eddie: Did Bob Siman's animals show plaques or neurological symptoms?
Eddie Koo: Bob Siman's animals were quite normal. But he recently crossed them to APP knock-in swedish animals and plaques formed early. I would be curious to see what they show when the analysis is completed.
Luciano: Yes. It would also be interesting to see if they now show more sensitivity to apoptosis.
June Kinoshita: Are you looking in human AD brain tissue for these fragments, or are they too transient to be seen postmortem?
Luciano: We reported in our JAD paper the isolation of AID-like peptides from the brains of normal and sporadic AD patients. Postmortem analysis is always tricky. It is difficult to distinguish between cause and effect, that is, pathogenetic mechanisms versus protective responses of the organism
Soshi: If C-terminal fragments are toxic, then why is the Swedish family with a ten-fold increase in gamma secretase ok until there are amyloid deposits? Data seem to support amyloid deposits.
Eddie Koo: But in the swedish mutation, 40 and 42 ratio is the same. So I assume Luciano can argue that there is no preferential increase in the 42 AID form.
Luciano: Soshi, they don't show symptoms till enough neurons are eliminated.
Eddie Koo: Others would argue that it is synapse loss, which precedes neurons loss, that is the early and more important substrate for clinical symptoms.
Soshi: Dr. D'Adamio, where in the cell is GFP AID fragment found?
Luciano: We saw AID everywhere---in all cell compartments.
Ben: Luciano - how might you relate the AID to early synaptic loss?
Luciano: Early synaptic loss could be mediated by a local activation of apoptotic mechanisms
Eddie Koo: With regard to postmortem brain, Andrea LeBlanc reported in D.C. that she sees more caspase activation in AD (I think caspase 6), as we showed with caspase 9. But again, there's smoke but no gun.
Ben: I guess the critical question is developing the right model system.
Luciano: Yes, Ben, you are right.
Eddie Koo: Ben, got any ideas which is the right model?
Luciano: The right model? That's a tricky question. AD is defined by the presence of plaques. Therefore, if you generate the symptoms of AD but not the plaques: would that be considered a relevant AD model ? (I think it should).
Ben: Eddie or Luciano, any thoughts about the relevence of that NGF deprivation model? There certainly would be apoptosis there.
Luciano: Yes, and as we showed together, Ben, it requires presenilins and APP.
Eddie Koo: Gene Johnson has certainly developed that model elegantly. But I don't know whether you can extend what happens in the sympathetic ganglia to the CNS. I don't know how much has been examined in the basal forebrain.
Ben: I guess the thing that strikes me in favor of Aβ is that in disease after disease, aggregation is the major problem.
Luciano: Are you referring to the Danish and British dementia ?
Ben: BDI, Huntington's, Parkinson's, ALS, etc. So MANY diseases have aggregates.
Luciano: Yes, but in which of these diseases have the aggregates been shown to be the cause of the disease ?
Ben: In Huntington's, the aggregate does not cause the disease, but the micro-aggregate (oligomer) seems to bind caspases. In PD, the aggregate is associated with death of dopaminergic neurons.
June Kinoshita: I thought the evidence in Huntington's is pretty strong, at least based on the reversal of symptoms in the mouse models when the expression of the mutant huntingtin is turned off and the inclusions go away.
Soshi: World Alzheimer Congress meeting data suggest that Aβ vaccine makes improvement in memory testing but no effect on Aβ levels - just Aβ aggregation. Please comment.
Ben: The only way to PROVE causation is to induce and reverse the aggregate. Soshi's comment is very appropriate.
Eddie Koo: In this regard, Lennart Mucke's recent paper comparing the APP and APP 717 mutant is quite informative. His group showed that synaptophysin loss can occur without plaques. It's how much 42 that's around. One can also interpret this in favor of Lucian's AID. Back to Ben's earlier statement, there is at least this instance where plaque aggregates are not so abundant.
Luciano: Soshi: In my opinion, there are several possibilities: for example, the polyclonal antibody response may recognize membrane bound APP and affect APP signalling through its cytoplasmic tail.
Ben: Luciano - the membrane bound APP would be intracellular.
June Kinoshita: {public msg} There is also the work from Dean Hartley and Bill Klein suggesting that non-aggregated forms of Aβ may be important.
Soshi: Is Mucke mouse a good model? Mouse already a stupid animal
Eddie Koo: At least in that paper, the stupidity of mice was not an issue.
Ben:Perhaps it was the stupidity of the reviewers!
June Kinoshita laughs hysterically
Luciano: Ours and Eddie's data show toxicity of the c-terminal region of APP. They also show that gamma secretase generates toxic intracellular peptides
Luciano: It is difficult to assume these peptides have no role in AD development.
Luciano: Ben: There is some APP at the cell surface as well.
Ben: Yes, but not the majority. OK Luciano - here is the question. What would constitute proof for AID in your mind?
Luciano: Ben: I think the development of an animal model in which you recapitulate symptoms, intracellular paired helical filaments and neuronal loss.
Ben: Don't the symptoms include plaques? Guest 3, will you identify yourself?
Eddie Koo: Guest 3 must be deep throat.
June Kinoshita: A spy from the NIH?
Ben: Must be Harold Varmus...........Hi Harold!!
Luciano: Behavioral symptoms. Plaques are histopathology. Harold has followed me to New York!!
Ben: But many things mimic the behavioral symptoms. A hippocampal lesion will give that. Even in Harold in NYC.
June Kinoshita: Poor Harold!
June Kinoshita: What about subtler aspects of neuronal dysfunction that precede neuronal death?
Luciano: June, neuronal dysfunction may be the consequence of activation of cell death in peripheral extensions of neurons. Also, it might be the consequence of attempts of cells to block apoptosis.
Eddie Koo: Didn't Dennis Dickson and the Mayo group report the NFT (tau mutant) crossed with the APP mice develop both tangles and palques? I seem to recall something about his saying APP increases the tau pathology but I can't remember precisely. Too much booze at the WAC
Ben: Hmm.....don't remember, but sounds believable. My sense is that many groups are seeing associated memory deficits.
Eddie Koo: Luciano, do you have any idea as to how AID works? What does AID activate? Your bcl-2 effects are similar to ours with C31.
Luciano: We have found several AID interacting proteins. Some of those are good candidates as mediators of AID induced cell death.
Eddie Koo: Luciano, are these interacting proteins different to what's been reported to bind the APP C-terminus? There are a bunch of them, some already hypothesized to be associated with increased cell death, like the G proteins.
Luciano:Yes. We have some new interesting candidates.
Eddie Koo: I need a deep throat in Luciano's lab.
Eddie Koo grins evilly
Luciano: It's the guy who is typing !
mcphie enters
Eddie Koo: Dr. McPhie, do you have any thoughts about this issue.? Rachael [Neve] obviously has worked with C100 for sometime.
June Kinoshita: Donna might want to comment on C-terminus-binding proteins as well.
mcphie leaves
June Kinoshita: Woops, looks like Donna is having problems with her system too.
Luciano: I think another important issue is: how is Aβ and AID generation (i.e. gamma secretase activity) regulated?
Ben: Yes, separate from whether Aβ or AID are the cause of AD, abnormal regulation must be somehow connected to the aging process.
June Kinoshita: So what enzyme cleaves the AID (C59) fragment to make C31?
Luciano: Caspases
Eddie Koo: The assumption from our data is caspase 8 and caspase 9 are required. We don't know which is the actual caspase.
Luciano: This is an important distinction because AID toxicity would imply gamma secretase as a regulator of programmed cell death. Caspases that generate C31 are already known to play a pivotal role in apoptosis.
Ben: Luciano, since PS2 is more closely connected to apoptosis - perhaps PS2 is more important for generating AID during proapoptotic conditions, and PS1 is more important under basal conditions. What do you think?
Luciano: Ben: This is an interesting possibility.
Ben: You should look at AID generation in cells lacking PS1 or cells lacking PS2.
Eddie Koo: So Luciano, have you done this experiment: express C99 in the setting of gamma-secretase inhibitor?
Luciano: We have been unable to obtain gamma secretase inhibitors from Merck. I am sure that Michael Wolfe will be kind enough to give us the reagents!
Eddie Koo: Ironically, in most if not all the studies with toxicity associated with APP C-terminus, none of the results are mutually exclusive. They could all be operating to some degree.
June Kinoshita: I wish Donna were here. I believe she and Rachael have found in their C100 cell cultures that gamma secretase inhibitors led to increased cell death.
Eddie Koo: I own no Merck stocks so you'll have to ask Mike and Dennis [Selkoe]. I am under the impression that Mike's inhibitors are also somewhat toxic.
Luciano: The important thing is that, initially, we cloned a C-terminal fragment of PS2 (ALG-3) as an inhibitor of fas-induced cell death. Now, we know that ALG-3 blocks gamma secretase activity and that's why it inihibits cell death
Ben: I have to run off to another meeting. Ciao!!
Ben bows gracefully
June Kinoshita: Thanks so much, Ben!
June Kinoshita: Any closing remarks?
Eddie Koo: It will be fun to see how the C-terminal toxicity plays out.
Luciano: Yes, June, I think the field should give more space to alternative hypotheses and possibilities. It is difficult to sell at any level data that does not directly supports the Aβ hypothesis and that is unfortunate.
Ben: Hmm.....No great wisdom. I really think the challenge, as always, is to examine the issues in a way that can exclude possibilities (e.g., Aβ toxicity or AID toxicity).
Luciano: I think we have to understand the mechanisms by which AID induces cell death. Second, we need to understand the mechanisms that regulate C99 processing and gamma secretase activity.
June Kinoshita: Thank you for participating today.
Eddie Koo: Gotta run, thanks for organizing this, June and Ben. And to Luciano for an interesting paper.
Ben: Eddie - I agree. Initially I was very enamored with apoptosis, but now I am more enamored with the idea of rust (e.g., cars die because of rust, etc). Ciao!
Luciano: Thanks to everyone for an interesting discussion !!
Ben: Thank you June and thanks Luciano and Eddie and Guest 3...........by Harold!
Luciano: Ben, the rust is due to oxidation.
Background
1T-cell apoptosis Unit, Laboratory of Cellular and Molecular Immunology, NIAID, National Institutes of Health, Bethesda, Maryland 20892; 2Section of Pharmacology and Neuroscience, IST, CBA and Dept. of Oncology Univ. of Genova, Genova, Italy; 3Laboratory of Immunology, NIAID, National Institutes of Health, Bethesda, Maryland 20892, 4Cytos Biotechnology AG / ETH Zurich, Wagistrasse 21, CH-8952, Zurich-Schlieren, Switzerland; 5Istituto di Anatomia Umana and Dipartimento di Neuroscienze, Universita' di Genova, via De Toni 10, 16132 Genova, Italy; 6Present address, Albert Einstein College of Medicine, Dept. of Microbiology & Immunology, 1300 Morris Park Avenue, Bronx, N.Y. 10461
*B.P. and L.P. have equally contributed to this work.
Address correspondence to Luciano D'Adamio, Albert Einstein College of Medicine, Dept. of Microbiology & Immunology, 1300 Morris Park Avenue, Bronx, N.Y. 10461
E-mail: ldadamio@aecom.yu.edu
Abstract
The b-amyloid precursor protein (APP) is sequentially processed by β- and γ- secretases to generate the Aβ peptide. The biochemical path leading to Aβ formation has been extensively studied since extracellular aggregates of amyloidogenic forms of Aβ peptide (Aβ42) are considered the culprit of Alzheimer's Disease. Aside from its pathological relevance, the biological role of APP proteolysis is unknown. Although never previously described, cleavage of APP by γ-secretase should release, together with Ab, a COOH-terminal APP intracellular domain, herein termed AID. We have now identified AID-like peptides in brain tissue of normal control and patients with sporadic Alzheimer's disease and demonstrate that AID acts as a positive regulator of apoptosis. Thus, overproduction of AID, may add to the toxic effect of Aβ42 aggregates and further accelerate neurodegeneration.
Alzheimer's disease (AD) is believed to be caused by extracellular deposition of amyloidogenic forms of Aβ peptide (Aβ42) (1, 2). Aβ derives from cleavage of APP by β- and γ-secretase (3) (Fig. 1A, upper panels). This hypothesis of AD pathogenesis, known as "amyloid hypothesis", has found further support by the identification of the three genes linked to familial forms of AD (FAD). The first discovered was APP, the protein from which Aβ is derived (4). The others are presenilin-1 (PS1) and -2 (PS2), two highly homologous proteins that are required for γ-secretase activity and might indeed be the γ-secretase (5, 6, 7, 8, 9). Of more importance, presenilins and APP point mutations found in FAD patients augment APP processing and the formation of amyloidogenic Aβ (1, 2, 10, 11).
Extensive evidence has also supported a role for presenilins and APP in programmed cell death (PCD). A dominant negative PS2 fragment, named ALG-3, was shown to inhibit apoptosis (12). This COOH-terminal PS2 fragment contains the second aspartate residue that is essential for γ-secretase activity (9) and codes for a dominant negative repressor of γ-secretase activity (13). Depletion of PS2 protein levels by antisense RNA has been shown to protect cells against death (14). Conversely, overexpression of presenilins increased apoptosis (14). Moreover, FAD-associated mutations in presenilins and APP enhanced the pro-apoptotic activity of these molecules (14, 15, 16). Lastly, apoptosis induced by APP requires Presenilins (14). Together, these data suggest an alternative model for the pathogenesis of AD. According to this hypothesis, neurodegeneration in AD is facilitated by enhanced susceptibility of neurons to apoptotic stimuli.
The "amyloid" and "apoptotic" theories need not be mutually exclusive. An attractive possibility is that APP processing may generate peptides that regulate PCD. This hypothesis provides a unifying model of these two apparently conflicting theories of Alzheimer's pathogenesis and is supported by the following findings. Conditions that increase the generation of the amyloidogenic form of Aβ, such as those with Alzheimer's mutation in presenilins and APP, also promote cell death. Conversely, circumstances that inhibit apoptosis, such as overexpression of ALG-3, also repress γ-secretase activity (13).
In this paper we show that the COOH-terminal APP intracellular domain, herein termed AID, liberated after cleavage of APP by γ-secretase acts as a positive regulator of apoptosis. Thus, overproduction of AID, as in AD, might cause the neurodegeneration process observed in Alzheimer's patients.
Experimental procedures.
Antibodies, constructs and mutagenesi.
The C7 rabbit polyclonal antiserum, raised to a synthetic polypeptide of APP corresponding to amino acids 751-770, was kindly donated by Dr. Dennis Selkoe. The 6E10 (anti-APP) and C10 (anti-PARP) mouse monoclonal antibodies were purchased from Senetek and Enzyme System (Dublin, CA), respectively. The monoclonal antibody 718-770/Jonas was purchased from Roche Molecular Biochemicals. The mouse monoclonal antibody anti-caspase-8 (Pharmingen, San Diego, CA) and the rabbit polyclonal antiserum against caspase-6 (Upstate Biotecnology, Lake Placid, NY) are both specific for the pro-domains of caspase-8 and 6 and do not recognize the active form. cDNA coding for the various human APP fragments were obtained by PCR and cloned into either pcDNA3 (Invitrogen, San Diego, CA) or pEGF-N1 (Clonethec, Palo Alto, CA). The APP-Sw APPD664N, AID57mut and AID59mut constructs were obtained by site-directed mutagenesis as suggested by the manufacturer's (CLONTECH, Palo Alto, CA). The identity of each construct was confirmed by automatic sequencing using an ABI Prism 377 (Perkin Elmer, Foster City, CA).
Cell lines and transfection procedures
Jurkat T-cells were transfected with a BTX electroporator with 30 mg of DNA and a setting of 1050 mF, 250 V and 72 W. Transfection efficiency was assessed using a GFP vector and ranged between 65 and 80%. One hr after transfection, cells dead because of the electroporation were removed by Ficoll-Paque (Pharmacia, Piscataway, NJ) centrifugation. In some experiments, the compound 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (5(6) (CFDA, SE) (Molecular Probes Inc, Eugene, OR), a green fluorescent dye, was added prior to transfections to mark a population of cells. Sub confluent HeLa or MCF7 cells were transfected with 1 mg of the indicated plasmids with FuGene 6 (Roche Molecular Biochemicals).
Apoptosis Studies
Cell death in Jurkat cells was assessed by either measuring the DNA content of isolated nuclei or by staining cells with annexin V-PE (R&D System, Minneapolis, MN) as indicated by the manufacturer. Samples were analyzed with a FACScan (Becton Dickinson, San Jose, CA). For HeLa cells, 16 hours later Hoechst 33342 was added to stain nuclei and cells were fixed with 3% paraformaldehyde and examined by fluorescence microscopy. Viable and apoptotic cells were enumerated by cell morphology and confirmed by examining nuclear morphology. At least 100 cells in each experimental group were counted in duplicate. Photomicrographs were taken at 630x magnification. When indicated, 50 mM of the irreversible caspase inhibitor ZVAD-fmk (Enzyme System, Livermore, CA) was used to block caspase activity.
Immunoprecipitation, Western blot analysis and caspase activity assays
For immunoprecipitation of metabolically labeled proteins, cells were incubated with 300mCi/ml of 35S-labeled methionine (Amersham, Arlington Heights, IL) for 4 hr. Cells were analyzed by immunoprecipitation as described previously. Western blots of total cell lysates were developed using the SuperSignal system (Pierce, Rockford, IL). For caspase activity assay, cells were lysed in 25 mM HEPES pH 7.5, 50 mM ß-mercaptoethanol, and 0.1% CHAPS and 30 µg of proteins were used in the enzymatic assay in 500 µl of AFC buffer (50 mM HEPES pH 7.5, 1% sucrose, 0.1% CHAPS), 1 mM DTT, and 50 µM ZDEVD-AFC (Enzyme System). All enzymatic reactions were carried out at 25°C for 2 h. Release of the fluorescent group was measured with a Perkin-Elmer LS-5B luminescence spectrometer at an excitation wavelength of 400 nm and an emission wavelength of 505 nm.
MALDI (Matrix-Assisted Laser Desorption Ionization) Mass Spectroscopy (MS)
Human brain samples were homogenized in a TBS buffer containing 1% of Triton X100. After a preclearing step with 40ml/ml of protein G-agarose, the extracted proteins were immunoprecipitated with the monoclonal antibody 643-695 /Jonas (10mg/ml) for 3 hours at 4C. Then 10ml of magnetic beads (Dynal, A.S. Oslo, Norway) covalently coupled with anti-mouse IgG were added to the sample and rocked for 1 hour in the same conditions. The magnetic beads were collected, washed twice with RIPA 1X, twice with ddH2O and suspended in 10 ml of ddH2O. Two ml of this slurry were then incubated briefly with 2 ml of the matrix: a-cyano-4-hydroxycinnamic acid (10mg/ml), (Aldrich Chem.Co. MI) dissolved in HCl 0.05M acetonitrile: isopropanol 5:1.5. One ml of the incubation mixture was placed on the sample plate with 1 ml of the matrix solution, evaporated at room temperature and then analyzed. Similar results were achieved using the 3,5-dimethoxy-4-hydroxycinnamic acid as matrix with or without formic acid in the matrix dilution. The analysis was performed in linear positive mode and a minimum of 100 scans was averaged.
Results and Discussion
To investigate the role of γ-secretase activity and APP processing in PCD, we initially studied cell death induced by Fas-associated death domain protein (FADD) (12). Transfection of FADD induced PCD in a dose- and temporal-dependent manner (Fig. 2A). While APP alone had negligible consequences, it augmented apoptosis triggered by FADD (3 mg) (Fig. 2A) and induced significant cell death when cotransfected with non-toxic doses of FADD (0.3 and 1 mg) (Fig. 2A). Assessing the cleavage of poly [ADR-ribose] polymerase (PARP) by cell death protease known as caspases (18) also corroborated these results. By 8 hrs, PARP was completely processed in cells transfected with the combination of APP and FADD (3 mg) as compared to approximately 60% cleavage in cells expressing FADD alone (Fig. 2B).
APP is first cleaved by β-secretase, giving rise to C99 (Fig. 1A, upper left panel). Alternatively, APP can be cleaved by a-secretase within the Aβ domain, generating a COOH-terminal membrane bound molecule of 83 amino acids (C83) (Fig. 1A, lower left panel). Processing of C99 and C83 fragments by the γ-secretase results in the release and secretion of Aβ and P3, respectively (19, 20). Concomitantly, a putative intracellular product that we referred to as APP Intracellular Domain (AID) should be generated (Fig. 1A, upper and lower right panels). Such a peptide has so far never been described. We asked whether these processed intermediates of APP were responsible for the apoptotic phenotype observed above. Constructs encoding for C99 and C83 were transfected either alone or with FADD. Neither C83 nor C99 induced PCD when expressed alone and only C99 synergized with FADD in inducing apoptosis (Fig. 2C and D). Interestingly, we observed the appearance of a shorter COOH-terminal APP fragment in C99 transfected cells, whose pattern of immunoreactivity and molecular weight was consistent with that of AID (Fig. 2E and F, left panel). This fragment was absent in C83 transfected cells suggesting that C99 is a better γ-secretase substrate than C83. To further address this question, cells were transfected with either wild type APP or the Swedish APP (APP-Sw) mutant (1, 2.). This FAD-associated mutant is more efficiently processed by b-secretase giving rise to more C99 than wild type APP (Fig. 2F, right panel). Consistent with our hypothesis, APP-Sw is more effectively degraded to AID polypeptides (Fig. 2F, right panel) and possesses stronger pro-apoptotic activity (not shown) than the wild type protein. Together, these data suggest a correlation between the strength of the apoptotic signal and the processivity of APP by γ-secretase.
The above results are compatible with the hypothesis that processing of C99 by γ-secretase can produce APP fragment(s) with pro-apoptotic functions. We therefore investigated whether one or both of the C99-derived fragments, Aβ and AID, mediate the observed effect on PCD. As a large fraction of Aβ is secreted upon production, we first tested whether FADD-induced cell death was increased by the secretion of Aβ. To address this, Jurkat cells were either labeled with the green fluorescent dye, CFSE, and cotransfected with FADD and C99 (CFSE+) or remained unlabeled and transfected with FADD only (CFSE-). The two populations were mixed immediately following transfection and assessed for PCD. If the synergistic effect on apoptosis was a consequence of Aβ secretion, then equivalent levels of cell death should be observed in both CFSE+ and CFSE- populations. Regardless of cell ratio, apoptosis was consistently observed in ~55% and ~35% of the CFSE+ and the CFSE- cells, respectively (Fig. 3A). These results indicate that secreted Aβ does not facilitate FADD-induced apoptosis. As an alternative approach, synthetic Aβ40 or Ab42 was directly added to Jurkat cells transfected with either vector control or FADD. Our results show that the addition of Aβ in the range of 5-10 mM did not reproduce the observed synergistic effects (Fig. 3B). Finally, as a further attempt to investigate whether Aβ synergizes with FADD, we transfected Jurkat cells with a construct that encodes for APPNcas. APPNcas represents the NH2-terminal fragment of APP generated by caspase-6 cleavage (Fig. 1B) (21, 22, 23, 24) and has been shown to generate higher levels of Aβ than full-length APP (24). In agreement with the above studies, FADD-induced apoptosis was not enhanced by APPNcas (Fig. 2C). Subsequently, we proceeded to test whether the pro-apoptotic function of C99 was mediated by its cytoplasmic tail, the putative AID peptide. To this end, we transfected a construct encoding for AID into Jurkat cells either alone or with FADD and cell death was measured both by DNA fragmentation (Fig. 2C) and PARP cleavage (Fig. 2D, right panel). Consistently, we observed that AID acted as a stronger inducer of FADD-meditated apoptosis as compared to both APP and C99. Thus, the synergistic effect of APP does not correlate with Aβ production, but is rather mediated by the APP COOH-terminal tail.
Although enhanced cell death by AID required FADD in Jurkat cells, we sought to determine whether overexpression of AID alone could trigger PCD in other cell lines. HeLa and MCF7 cells were transfected with plasmids encoding various APP-derived fragments fused to green fluorescent protein (GFP) to directly visualize transfected cells. While overexpression of either APP (Fig. 4 and 5B) or APPNcas (not shown) did not affect cell viability, transfection of AID in either cell line consistently generated elevated levels of cell death (25-35%) as defined by cell shrinkage and nuclear condensation (Fig. 4 and 5B). From these studies, three lines of evidence demonstrate that AID induces an apoptotic form of cell death. First, overexpression of AID induced activation of caspases (Fig. 4C and D), which are cysteine proteases that implement PCD (18). Second, activation of caspases is required for the execution of cell death since the caspase inhibitors zVAD-fmk, Crma, p35 and MC159 blocked AID-induced apoptosis (Fig. 4A and B). Lastly, the anti-apoptotic protein Bcl-XL (Fig. 5B), a Bcl-2 family member, also inhibited AID-induced cell death.
C99 can be cleaved by the γ-secretase at two distinct positions to generate either Aβ40 or Ab42. The corresponding AID fragments would comprise either the 58 (AID59) or 56 (AID57) COOH-terminal amino acid of APP, respectively (Fig. 5A). FAD mutations in APP and presenilins all result in a shift in metabolism of APP such that more Aβ42 is produced. Consequently, increased amounts of AID57 will be released in the cytosol. If the shorter AID57 peptides were more toxic than the longer form, this could explain why APP and presenilins FAD mutants have enhanced pro-apoptotic activity than the corresponding wild type. To address this question, HeLa and MCF-7 cells were transfected with vectors expressing either AID59 or AID57 and analyzed for cell death. Strikingly, our data revealed that AID57 was significantly more potent than AID59 in inducing PCD (Fig. 5B). Moreover, in a mouse motor neuronal cell line (MN-1) (25), similar results were observed. That is, AID57 was more effective than AID59 in promoting apoptosis.
Interestingly, in all three cells lines, overexpression of APPCcas, a 31 amino acid COOH-terminal fragment of APP released by caspase-6 cleavage (Fig. 1B), was non-toxic. These results are contrary to those recently published (26), which demonstrated that C31, a COOH-terminal polypeptide corresponding to APPCcas, acts as an amplifier of PCD. We further addressed this discrepancy by asking whether disruption of the caspase cleavage site within the cytoplasmic tail of APP abrogates its inducing affect. A substitution of an aspartic acid residue for an asparagine was introduced at position 664 in APP (APPD664N), AID59 (AID59mut) and AID57 (AID57mut), and subsequently tested for cell death in Jurkat, HeLa and MCF-7 cells. In Jurkat cells, overexpression of either APP full-length or AID57-containing mutants were not compromised in their ability to augment FADD-induced apoptosis (Fig 5C). By contrast and in accordance with the above data, APPCcas was ineffective in amplifying the effects of FADD on cell death. Also, comparable levels of PCD were observed in HeLa cells bearing either AID57 or AID57mut (Fig. 5C). Lastly, both AID59 and AID59mut activated cell death in either HeLa or MCF-7 cells (Fig. 5C). Together, these results support a prerequisite for γ-secretase-mediated release of AID for induction of apoptosis, and, moreover, argue against the requirement of further processing.
Although the knowledge available on APP processing argues that one AID molecule must be produced for every Aβ peptide released (Fig. 1A), AID peptides have never been described previously. To substantiate the physiological and pathological significance of our findings, we investigated whether AID-like peptides are present in post-mortem sporadic AD and normal brain tissues (25-maldi) (27, 28, 29). As shown in Fig. 5D, four AID peptides were isolated from these tissues. These peptides were identified by MALDI-MS sequence analysis as AID fragments that undergo further proteolysis in vivo at both the NH2- and COOH-terminus.
Here we show that a natural product of γ-secretase cleavage, the cytoplasmic tail of APP, is a positive regulator of PCD. While in Jurkat cells it facilitates FADD-dependent apoptosis, AID directly triggers PCD in HeLa, MCF7 and MN-1 cells. Whether this difference is cell-type dependent it remains to be investigated. These data suggest that proteolysis of APP by secretases tunes the susceptibility of cells to apoptosis. In this scenario, presenilins might facilitate PCD by promoting cleavage of APP by the γ-secretase, thus governing the amount of AID generated. The biological and pathological relevance of this model is endorsed by the discovery that AID peptides are detected in normal and sporadic AD brain. The functional consequences of APP processing described above resembles that of Notch and Ire1, two other proteins whose processing is controlled by presenilins (30); release of the intracellular domain of Notch and Ire1 by cleavage within the transmembrane region results in downstream effector function.
Could these findings be applied to the pathogenesis of Alzheimer's disease? Our studies suggest that overproduction of AID, and especially the shorter AID57 peptide, makes cells more sensitive to apoptotic stimuli. This may add to the toxic burden caused by the amyloidogenic plaques and by Aβ released in the endoplasmic reticulum (31), further accelerating the neurodegenerative process observed in the brain of Alzheimer's patients.
Figure legend
Figure 1
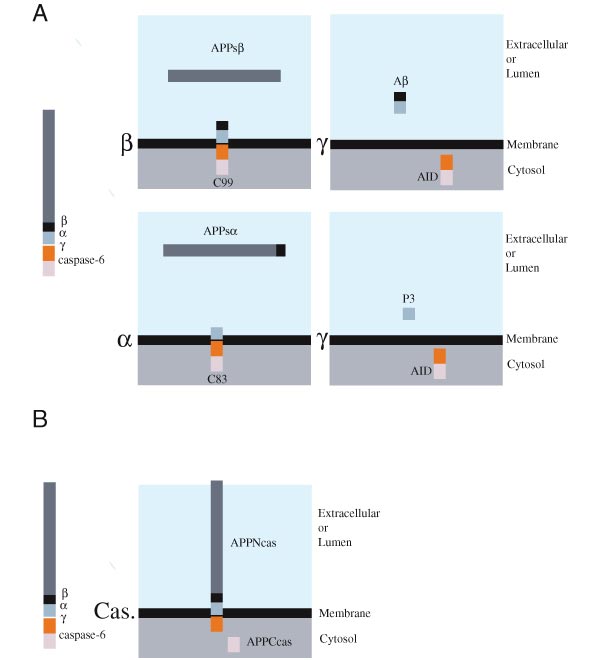 Endoproteolysis of APP by secretases and caspase-6. (A) APP processing occurs by a series of cleavage events that lead to the formation of Aβ and AID. To initiate the generation of Aβ, APP is cleaved by β-secretase at the NH2-terminus of Aβ to release APPsb and C99, a membrane-bound fragment (upper left panel). Alternatively, cleavage by the α-secretase results in the generation of a large NH2-terminal fragment, APPsa, and a membrane-bound COOH-terminal polypeptide, C83 (lower left panel). C99 and C83 can be further cleaved by γ-secretase to release and secrete Aβ or P3 peptide, respectively (upper and lower right panel, respectively). In both cases, a ~6 kDa cytosolic AID fragment should be generated. (B) APP cleavage by caspase-6 occurs at a caspase consensus sequence (VEVD) within the cytoplasmic tail of APP. This cleavage event results in the generation of APPNcas, an NH2-terminal membrane-associated fragment, and APPCcas, a cytosolic fragment of 31 amino acids.
Endoproteolysis of APP by secretases and caspase-6. (A) APP processing occurs by a series of cleavage events that lead to the formation of Aβ and AID. To initiate the generation of Aβ, APP is cleaved by β-secretase at the NH2-terminus of Aβ to release APPsb and C99, a membrane-bound fragment (upper left panel). Alternatively, cleavage by the α-secretase results in the generation of a large NH2-terminal fragment, APPsa, and a membrane-bound COOH-terminal polypeptide, C83 (lower left panel). C99 and C83 can be further cleaved by γ-secretase to release and secrete Aβ or P3 peptide, respectively (upper and lower right panel, respectively). In both cases, a ~6 kDa cytosolic AID fragment should be generated. (B) APP cleavage by caspase-6 occurs at a caspase consensus sequence (VEVD) within the cytoplasmic tail of APP. This cleavage event results in the generation of APPNcas, an NH2-terminal membrane-associated fragment, and APPCcas, a cytosolic fragment of 31 amino acids.
Figure 2
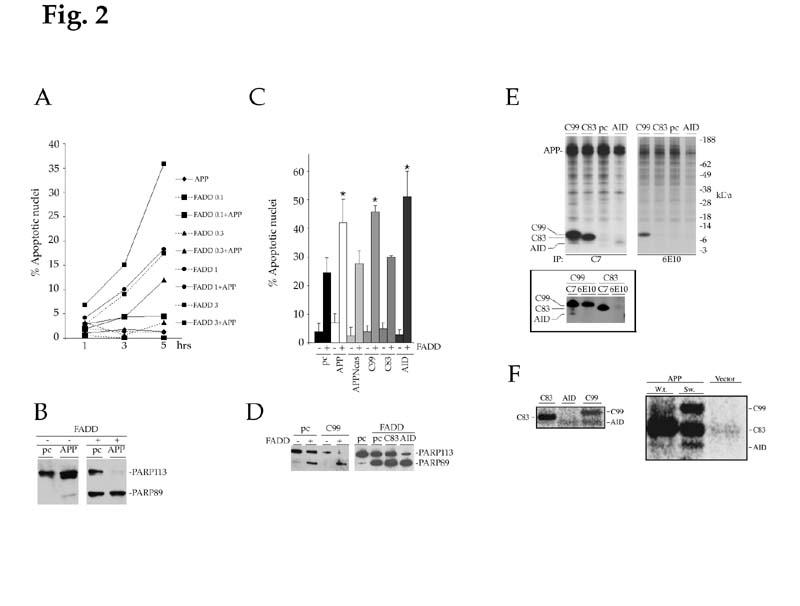 APP and its naturally processed derivatives augment FADD-induced PCD. (A) APP facilitates FADD-induced cell death. Jurkat cells were transfected with either APP (10 mg), FADD (0.1-3 mg) or in combination. At the indicated time points, apoptosis was measured by determining the percentage of nuclei undergoing DNA fragmentation. (B) FADD-induced PARP cleavage is increased by APP. 8 hrs post-transfection, Jurkat lysates were prepared from cells transfected with APP and/or FADD. Protein lysates (~7 mg/lane) were separated on a 4-12% PAGE, blotted and probed with an anti-PARP antibody. Note the complete cleavage of PARP (89 kDa) in cells cotransfected with APP and FADD. (D) Synergistic effects on FADD-induced PCD by APP-derived polypeptides. Jurkat cells were transfected with empty pcDNA3 vector (pc) or constructs encoding for various APP fragments (10 mg) either in the absence (-) or presence (+) of FADD (3 mg). The results represent the mean +/- S.D. of 5 independent experiments. Apoptosis was assessed 6 hrs post-transfection. A student's t-test was used to calculate the results and * indicates that P values were (D) C99 and AID augment FADD-induced PARP cleavage. Cell lysates were prepared from Jurkat cells transfected with pcDNA3 (pc) or C99 alone (-) or in combination with FADD (+) (left panel). In other experiments (right panel), lysates were prepared from control cells (pc) or cells transfected with FADD together with pcDNA3 (pc), C83 or AID. Samples were analyzed for PARP cleavage 8 hours post-transfection. C99 and AID increased the percentage of cellular PARP processed during FADD-induced apoptosis. Neither the vector control nor the various APP fragments alone induced PARP cleavage. (E) Cells were transfected with pcDNA3 (pc), C99, C83 or AID. Labeled proteins were immunoprecipitated with either 6E10 or C7 anti-APP antibodies. Since the epitope recognized by the anti-6E10 antibody is located between the β- and α- secretase sites of APP, only C99 was precipitated by this antiserum. Conversely, the C7 antiserum, specific for the COOH-terminal 20 amino acids of APP, precipitated all three APP-derived fragments. Interestingly, a polypeptide of molecular weight similar to AID was immunoprecipitated by C7 but not 6E10 antibodies in C99-transfected cells,
APP and its naturally processed derivatives augment FADD-induced PCD. (A) APP facilitates FADD-induced cell death. Jurkat cells were transfected with either APP (10 mg), FADD (0.1-3 mg) or in combination. At the indicated time points, apoptosis was measured by determining the percentage of nuclei undergoing DNA fragmentation. (B) FADD-induced PARP cleavage is increased by APP. 8 hrs post-transfection, Jurkat lysates were prepared from cells transfected with APP and/or FADD. Protein lysates (~7 mg/lane) were separated on a 4-12% PAGE, blotted and probed with an anti-PARP antibody. Note the complete cleavage of PARP (89 kDa) in cells cotransfected with APP and FADD. (D) Synergistic effects on FADD-induced PCD by APP-derived polypeptides. Jurkat cells were transfected with empty pcDNA3 vector (pc) or constructs encoding for various APP fragments (10 mg) either in the absence (-) or presence (+) of FADD (3 mg). The results represent the mean +/- S.D. of 5 independent experiments. Apoptosis was assessed 6 hrs post-transfection. A student's t-test was used to calculate the results and * indicates that P values were (D) C99 and AID augment FADD-induced PARP cleavage. Cell lysates were prepared from Jurkat cells transfected with pcDNA3 (pc) or C99 alone (-) or in combination with FADD (+) (left panel). In other experiments (right panel), lysates were prepared from control cells (pc) or cells transfected with FADD together with pcDNA3 (pc), C83 or AID. Samples were analyzed for PARP cleavage 8 hours post-transfection. C99 and AID increased the percentage of cellular PARP processed during FADD-induced apoptosis. Neither the vector control nor the various APP fragments alone induced PARP cleavage. (E) Cells were transfected with pcDNA3 (pc), C99, C83 or AID. Labeled proteins were immunoprecipitated with either 6E10 or C7 anti-APP antibodies. Since the epitope recognized by the anti-6E10 antibody is located between the β- and α- secretase sites of APP, only C99 was precipitated by this antiserum. Conversely, the C7 antiserum, specific for the COOH-terminal 20 amino acids of APP, precipitated all three APP-derived fragments. Interestingly, a polypeptide of molecular weight similar to AID was immunoprecipitated by C7 but not 6E10 antibodies in C99-transfected cells,
(F) Cells were transfected with C83, AID, C99 (left panel), APP wild type (W.t.), APP Swedish mutant (Sw.) or pcDNA3 (Vector) (right panel). Labeled proteins were immunoprecipitated with the C7 antiserum. While detectable levels of AID peptide were identified in C99 transfected cells, this fragment was not apparent in cells overexpressing C83 (Right panel). The APP-Sw mutant is processed at the b-secretase site more efficiently than W.t., APP, and consequently, generates higher levels of AID-like peptide.
Figure 3
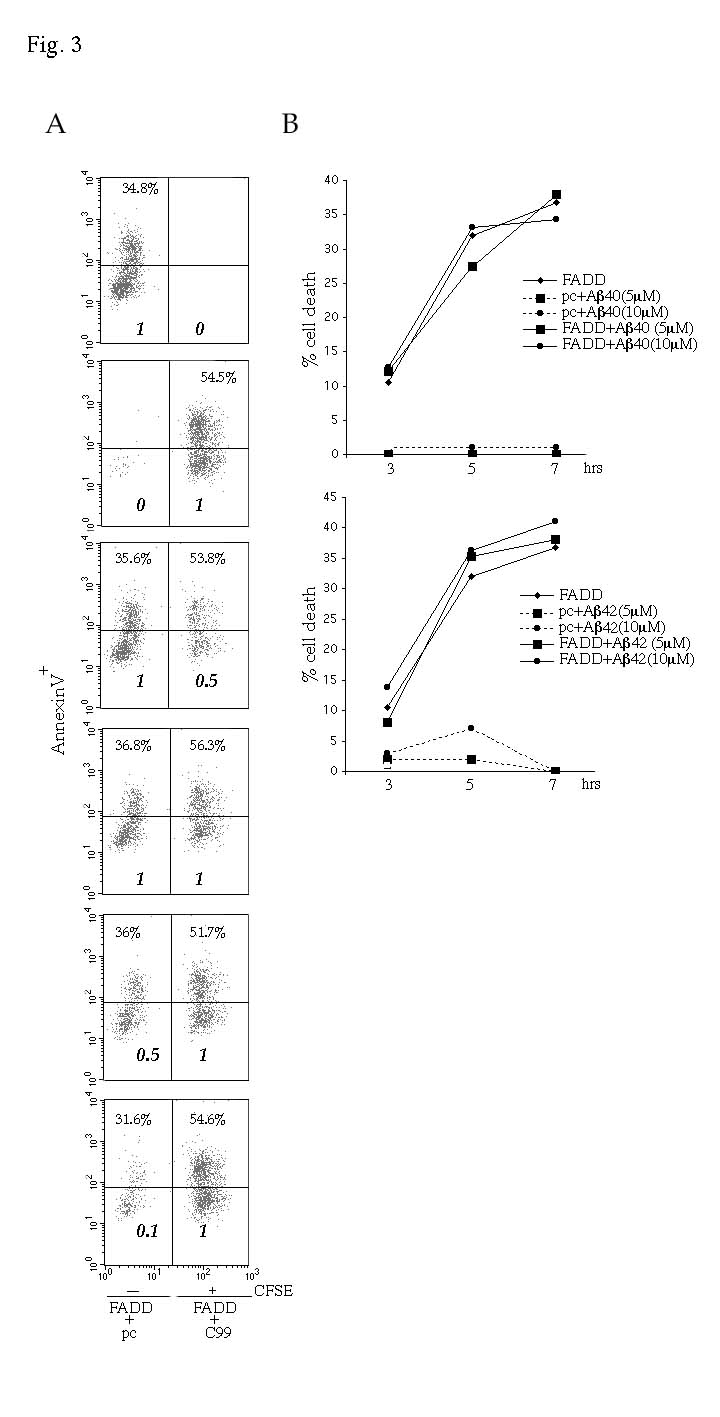 The synergistic effect of APP and its processed forms on FADD-induced PCD is not dependent on secreted Aβ. (A) Secreted Aβ derived from CFSE+ cells does not enhance FADD-induced PCD in CFSE- cells. Jurkat cells labeled with the green fluorescent dye ,CFSE, were transfected with C99 and FADD, while unlabeled cells were transfected with FADD only. Immediately following transfection, unlabeled and labeled cells were mixed according to the ratios indicated (lower left and right quadrants). Apoptosis in CFSE+ (left quadrants) and CFSE- (right quadrants) populations was measured 6 hrs post-transfection by determining the percentage of AnnexinV+ cells. This protein binds phosphatidylserines that are "flipped" and exposed on the cell surface during PCD. CFSE+ cells (~55%) consistently induced higher levels of cell death than CFSE- cells (~35%), indicating a cis and not a trans-effect. (B) Apoptosis induced by FADD is not enhanced by addition of exogenous Aβ40 or Aβ42. Aβ40 or Aβ42 peptides (5-10 mM) were added immediately following transfection of Jurkat cells with either pcDNA3 vector control (pc) or FADD (3 mg). Of note, higher concentrations of Aβ42 (25 mM) induced significant apoptosis in Jurkat cells, indicating that the peptide preparation was cytotoxic (data not shown).
The synergistic effect of APP and its processed forms on FADD-induced PCD is not dependent on secreted Aβ. (A) Secreted Aβ derived from CFSE+ cells does not enhance FADD-induced PCD in CFSE- cells. Jurkat cells labeled with the green fluorescent dye ,CFSE, were transfected with C99 and FADD, while unlabeled cells were transfected with FADD only. Immediately following transfection, unlabeled and labeled cells were mixed according to the ratios indicated (lower left and right quadrants). Apoptosis in CFSE+ (left quadrants) and CFSE- (right quadrants) populations was measured 6 hrs post-transfection by determining the percentage of AnnexinV+ cells. This protein binds phosphatidylserines that are "flipped" and exposed on the cell surface during PCD. CFSE+ cells (~55%) consistently induced higher levels of cell death than CFSE- cells (~35%), indicating a cis and not a trans-effect. (B) Apoptosis induced by FADD is not enhanced by addition of exogenous Aβ40 or Aβ42. Aβ40 or Aβ42 peptides (5-10 mM) were added immediately following transfection of Jurkat cells with either pcDNA3 vector control (pc) or FADD (3 mg). Of note, higher concentrations of Aβ42 (25 mM) induced significant apoptosis in Jurkat cells, indicating that the peptide preparation was cytotoxic (data not shown).
Figure 4
 AID induces apoptosis in HeLa cells. HeLa cells were transfected with 2 mg of mammalian expression vectors coding for APP-GFP, AID-GFP, FADD-GFP or GFP. Cells were stained with Hoechst H33258 24 hrs after transfection, and apoptotic nuclei were scored with an inverted fluorescence microscope. At least 100 cells/sample were counted for each data point. Percentages are the average of three independently transfected wells. Similar results were obtained in two other experiments. (A) The caspase inhibitor, Z-VAD-fmk, blocks cell death triggered by AID. Note that Z-VAD-fmk blocks AID-induced apoptosis as efficiently as FADD. (B) Viral-derived caspase inhibitors protect HeLa cells from AID-induced PCD. Cotransfection of AID with plasmids encoding either CrmA (blocks caspase1 and 8), p35 (blocks multiple caspases) or MC159 (also called v-flip; blocks caspase 8 and FADD) completely blocked the effects of AID. (C) Protein levels of caspase-6 and caspase-8 precursors (pro-Cas.8 and pro-Cas.6) are diminished in AID transfected cells. Equivalent amounts of protein were determined by blotting with anti-BNIP2. Note that similar affects were observed with FADD. (D) AID activates caspases as determined by in vitro cleavage of the fluorescent substrate DEVD-AFC. Note that the same cell lysates from (C) were used to monitor for DEVD-AFCase activity.
AID induces apoptosis in HeLa cells. HeLa cells were transfected with 2 mg of mammalian expression vectors coding for APP-GFP, AID-GFP, FADD-GFP or GFP. Cells were stained with Hoechst H33258 24 hrs after transfection, and apoptotic nuclei were scored with an inverted fluorescence microscope. At least 100 cells/sample were counted for each data point. Percentages are the average of three independently transfected wells. Similar results were obtained in two other experiments. (A) The caspase inhibitor, Z-VAD-fmk, blocks cell death triggered by AID. Note that Z-VAD-fmk blocks AID-induced apoptosis as efficiently as FADD. (B) Viral-derived caspase inhibitors protect HeLa cells from AID-induced PCD. Cotransfection of AID with plasmids encoding either CrmA (blocks caspase1 and 8), p35 (blocks multiple caspases) or MC159 (also called v-flip; blocks caspase 8 and FADD) completely blocked the effects of AID. (C) Protein levels of caspase-6 and caspase-8 precursors (pro-Cas.8 and pro-Cas.6) are diminished in AID transfected cells. Equivalent amounts of protein were determined by blotting with anti-BNIP2. Note that similar affects were observed with FADD. (D) AID activates caspases as determined by in vitro cleavage of the fluorescent substrate DEVD-AFC. Note that the same cell lysates from (C) were used to monitor for DEVD-AFCase activity.
Figure 5
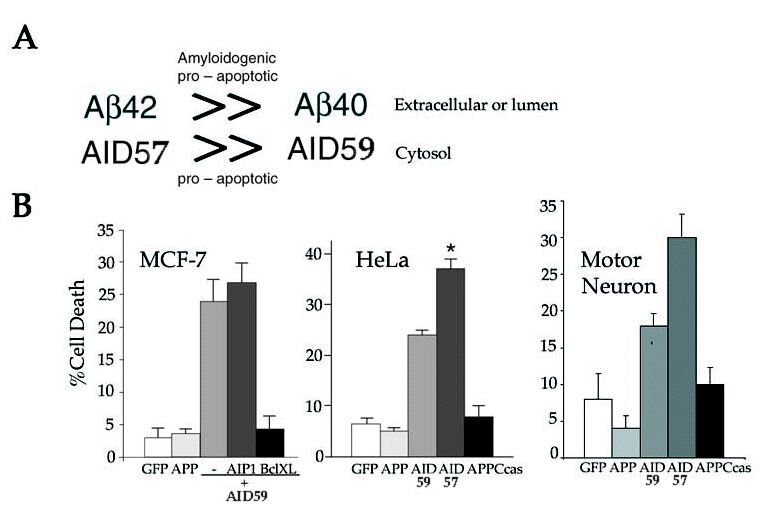
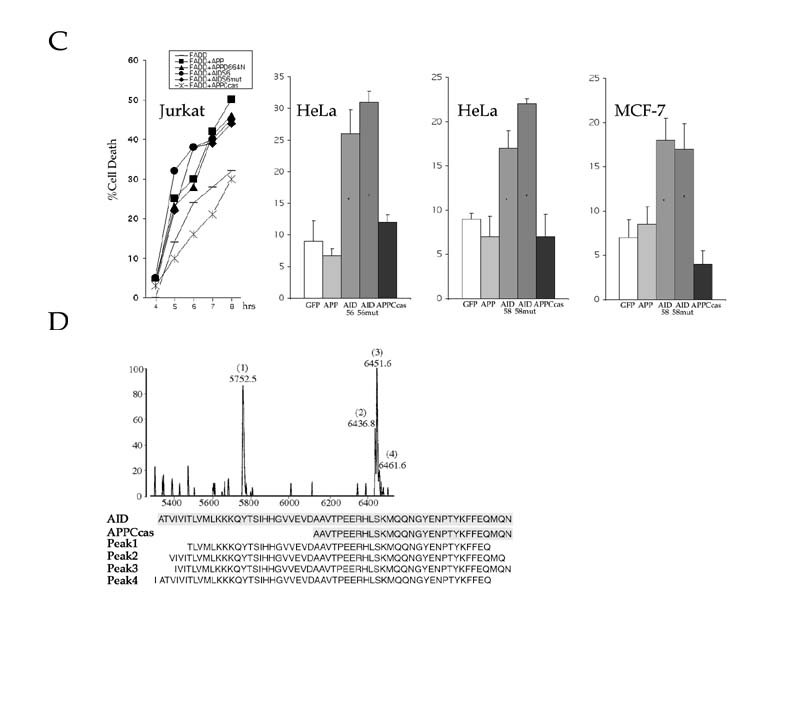 (A) γ-secretase cleavage of APP can occur at two different positions. A cut occurring between residues 637-638(indicated as β-40) gives rise to the short Aβ (Aβ40) and long AID (AID59). Conversely, cleavage after residue 639 (indicated as g-42) yields the longer Aβ (Aβ42) isoform and shorter AID (AID57) fragment (numbering is according to the 695 amino acid long APP isoform). FAD mutations preferentially increase cleavage after residue 639, which result in the production of the highly amyloidogenic Ab42 peptide. In this model, we postulate that the resulting AID57, is more damaging to cells than its longer AID59 counterpart. Thus, FAD mutations will result in overproduction of two APP-derived peptides that exert their neurotoxic action both intra- (AID57) and extracellularly (Aβ42). Aβ peptides could also exert a pro-apoptotic activity that requires caspase-12 in the endoplasmic reticulum (E.R.) compartment (37). (B) (left panel) AID-induced apoptosis in MCF-7 cells is inhibited by the anti-apoptotic protein Bcl-XL (data not shown for HeLa and MN-1). Expression of an unrelated control protein (AIP1) did not influence cell death. (Middle panel) AID57 induces significantly more apoptosis than AID59 in HeLa cells (data not shown for MCF-7) (*P (C) Disruption of the caspase cleavage site within the cytoplasmic tail of APP and AID does not impair the execution of cell death. APPD664N and AID57mut were transfected into Jurkat cells (left panel) either alone (data not shown) or with FADD and analyzed at the indicated time points for apoptosis. As compared to their non-mutant counterparts, APPD664N and AID57mut were no different in their ability to implement apoptosis. Conversely, overexpression of APPCcas exhibited negligible effects on FADD-induced cell death. Note that the vector control background was subtracted from each time point. In HeLa cells, overexpression of either AID57mut (middle left panel) or AID59mut (middle right panel) induced cell death to the same extent of their non-mutant counterparts, whereas APPCcas was incapable in producing such effects. Similar results were also obtained in MCF-7 cells (right panel) where overexpression of AID59mut displayed comparable levels of cell death to AID59. (D) AID-like peptides are present in normal and sporadic AD brain. Sequence analysis of four peptides (peak 1-4) immunoprecipitated by the Jonas monoclonal antibody, which are recognized on western blot by an anti-APP antiserum (not shown) are compared to the sequences of AID59 and APPCcas. In the experiment shown, a post-mortem brain tissue from a 72 years old AD patient was analyzed. These AID-like peptides were also found in the three other post-mortem brains that were examined. Two were from normal controls (45 and 51 years of age) and one other AD (65 years of age).
(A) γ-secretase cleavage of APP can occur at two different positions. A cut occurring between residues 637-638(indicated as β-40) gives rise to the short Aβ (Aβ40) and long AID (AID59). Conversely, cleavage after residue 639 (indicated as g-42) yields the longer Aβ (Aβ42) isoform and shorter AID (AID57) fragment (numbering is according to the 695 amino acid long APP isoform). FAD mutations preferentially increase cleavage after residue 639, which result in the production of the highly amyloidogenic Ab42 peptide. In this model, we postulate that the resulting AID57, is more damaging to cells than its longer AID59 counterpart. Thus, FAD mutations will result in overproduction of two APP-derived peptides that exert their neurotoxic action both intra- (AID57) and extracellularly (Aβ42). Aβ peptides could also exert a pro-apoptotic activity that requires caspase-12 in the endoplasmic reticulum (E.R.) compartment (37). (B) (left panel) AID-induced apoptosis in MCF-7 cells is inhibited by the anti-apoptotic protein Bcl-XL (data not shown for HeLa and MN-1). Expression of an unrelated control protein (AIP1) did not influence cell death. (Middle panel) AID57 induces significantly more apoptosis than AID59 in HeLa cells (data not shown for MCF-7) (*P (C) Disruption of the caspase cleavage site within the cytoplasmic tail of APP and AID does not impair the execution of cell death. APPD664N and AID57mut were transfected into Jurkat cells (left panel) either alone (data not shown) or with FADD and analyzed at the indicated time points for apoptosis. As compared to their non-mutant counterparts, APPD664N and AID57mut were no different in their ability to implement apoptosis. Conversely, overexpression of APPCcas exhibited negligible effects on FADD-induced cell death. Note that the vector control background was subtracted from each time point. In HeLa cells, overexpression of either AID57mut (middle left panel) or AID59mut (middle right panel) induced cell death to the same extent of their non-mutant counterparts, whereas APPCcas was incapable in producing such effects. Similar results were also obtained in MCF-7 cells (right panel) where overexpression of AID59mut displayed comparable levels of cell death to AID59. (D) AID-like peptides are present in normal and sporadic AD brain. Sequence analysis of four peptides (peak 1-4) immunoprecipitated by the Jonas monoclonal antibody, which are recognized on western blot by an anti-APP antiserum (not shown) are compared to the sequences of AID59 and APPCcas. In the experiment shown, a post-mortem brain tissue from a 72 years old AD patient was analyzed. These AID-like peptides were also found in the three other post-mortem brains that were examined. Two were from normal controls (45 and 51 years of age) and one other AD (65 years of age).
References
1 Price, D. L., and Sisodia, S. S. (1998) Annu. Rev. Neurosci. 21, 479-505.
2. Hardy, J. (1997) Trends Neurosci. 20, 154-159
3. Haass, C. and Selkoe, D.J. (1993) Cell 75,1039-1042
4. Goate, A., Chartier-Harlin, M.C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., Mant, R., Newton, P., Rooke, K., Roques, P., Talbot, C., Pericakvance, M., Roses, A., Williamson, R., Rossor, M., Owen, M., and Hardy, J. (1991) Nature 349, 704-706
5. Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., Tsuda, T., Mar, L., Foncin, J.-F., Bruni, A. C., Montesi, M. P., Sorbi, S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D., Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A. R., Haines, J. L., Pericak-Vance, M. A., Tanzi, R. E., Roses, A. D., Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995) Nature 375, 754-760
6. Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D. M., Oshima, J., Pettingell, W. H., Yu, C., Jondro, P. D., Schmidt, S. D., Wang, K., Crowley, A. C., Fu, Y.-H., Guenette, S. Y., Galas, D., Nemens, E., Wijsman, E. M., Bird, T. D., Schellenberg, G. D., and Tanzi, R. E. (1995) Science 269, 973-977
7. Rogaev, E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., Mar, L., Sorbl, S., Nacmias, B., Piacentini, S., Amaducci, L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995) Nature 376, 775-778
8. De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., Von Figura, K., and Van Leuven F. (1998) Nature 391, 387-390
9. Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999) Nature 398, 513-517.
10. Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C.-M., Perez-tur, J., Hutton, M., Buee, L., Harigaya, Y., Yager, D., Morgan, D., Gordon, M. N., Holcomb, L., Refolo, L., Zenk, B., Hardy, J., and Younkin, S. (1996) Nature 383, 710-713
11. Scheuner ,D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., Bird, T.D., Hardy, J., Hutton, M., Kukull, W., Larson, E., Levy-Lahad, E., Viitanen, M., Peskind, E., Poorkaj, P., Schellenberg, G., Tanzi, R., Wasco, W., Lannfelt, L., Selkoe, D., and Younkin, S. (1996) Nat. Med. 2, 710-713
12. Vito, P., Lacaná, E., and D'Adamio, L. (1996) Science 271, 521-525
13. Palacino, J. J., Berechid, B. E., Alexander, P., Eckman, C., Younkin, S., Nye, J. S., and Wolozin, B. (2000) J. Biol. Chem. 275, 215-222.
14. Wolozin, B., Iwasaki, K., Vito, P., Ganjei, J. K., Lacaná, E., Sunderland, T., Zhao, B., Kusiak, J. W., Wasco, W., and D'Adamio, L. (1996) Science 274, 1710-1713
15. Guo, Q., Sopher, B. L., Furukawa, K., Pham, D. G., Robinson, N., Martin, G. M., and Mattson, M. P. (1997) J. Neurosci. 17, 4212-4222
16. Yamatsuji, T., Okamoto, T., Takeda, S., Murayama, Y., Tanaka, N., and Nishimoto, I. (1996) EMBO J. 15, 498-509.
17. Chinnaiyan, A. M., O'Rourke K., Tewari, M. V., and Dixit, M. (1995) Cell 81, 505-513
18. Thornberry, N. A., Lazebnik, Y. (1998) Science 281, 1312-1313
19. Sinha, S., and Lieberburg, I. (1999) Proc. Natl. Acad. Sci. U. S. A. 96, 11049-11053
20. Haass, C., and De Strooper, B. (1999) Science 286, 916-917
21. Weidemann, A., Paliga, K., Durrwang, U., Reinhard, F.B.M., Schuckert, O., Evin, E., and Masters, C.L. (1999) J. Biol. Chem. 274, 5823-5829
22. Pellegrini, L., Passer, B. J., Tabaton, M., Ganjei, J. K., and D'Adamio, L. (1999) J. Biol. Chem. 274, 21011-21016
23. LeBlanc, A., Liu, H. , Goodyer, C., Bergeron, C., and Hammond, J. (1999) J. Biol. Chem. 274, 23426-23431
24. Gervais, F. G., Xu, D., Robertson, G. S., Vaillancourt, J. P., Zhu, Y., Huang, J., LeBlanc, A., Smith, D., Rigby, M., Shearman, M. S., Clarke, E.E., Zheng, H., Van Der Ploeg, L. H., Ruffolo, S. C., Thornberry, N. A., Xanthoudakis, S., Zamboni, R. J., Roy, S., and Nicholson, D. W. (1999) Cell 97, 395-403
25. Salazar-Grueso, E. F, Kim, S., and Kim, H. (1991) Neuroreport 2, 505-511
26. Lu, D. C., Rabizadeh, S., Chandra, S., Shayya, R. F., Ellerby, L.M., Ye, X., Salvesen, G. S., Koo, E.H., and Bredesen, D.E. (2000) Nature Med. 6, 397-404
27. De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J.S., Schroeter, E.H., Schrijvers, V., Wolfe, M.S., Ray W.J., Goate, A., and Kopan, R. (1998) Nature 398, 518-522
28. Struhl, G., and Greenwald, I. (1999) Nature 398, 522-525
29. Ye, Y., Lukinova, N., and Fortini, M. E. (1999) Nature 398, 525-529
30. Niwa, M., Sidrauski, C., Kaufman, R. J., and Walter, P. A. (1999) Cell 99, 691-699
31. Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, .J, Yankner, B. A., and Yuan, J. (2000) Nature 403, 98-103
References
Webinar Citations
Paper Citations
- García-Redondo A, Dols-Icardo O, Rojas-García R, Esteban-Pérez J, Cordero-Vázquez P, Muñoz-Blanco JL, Catalina I, González-Muñoz M, Varona L, Sarasola E, Povedano M, Sevilla T, Guerrero A, Pardo J, López de Munain A, Márquez-Infante C, de Rivera FJ, Pastor P, Jericó I, de Arcaya AÁ, Mora JS, Clarimón J, C9ORF72 Spanish Study Group, Gonzalo-Martínez JF, Juárez-Rufián A, Atencia G, Jiménez-Bautista R, Morán Y, Mascías J, Hernández-Barral M, Kapetanovic S, García-Barcina M, Alcalá C, Vela A, Ramírez-Ramos C, Galán L, Pérez-Tur J, Quintáns B, Sobrido MJ, Fernández-Torrón R, Poza JJ, Gorostidi A, Paradas C, Villoslada P, Larrodé P, Capablo JL, Pascual-Calvet J, Goñi M, Morgado Y, Guitart M, Moreno-Laguna S, Rueda A, Martín-Estefanía C, Cemillán C, Blesa R, Lleó A. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. 2013 Jan;34(1):79-82. Epub 2012 Oct 11 PubMed.
Other Citations
External Citations
Further Reading
Papers
- Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999 Feb 25;397(6721):698-701. PubMed.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.