Microglia Rely on Mixed Messages to Select Synapses for Destruction
Quick Links
Microglia may be the brain’s trash collectors, but when it comes to neurons, they are quite discerning, it turns out. At “Inflammation in Diseases of the Central Nervous System,” a Keystone symposium held January 25-30 in Taos, New Mexico, researchers reported that the cells interpret a combination of go/no-go signals to select synapses for disposal. Researchers raised the possibility that the signals go haywire in neurodegenerative diseases such as Alzheimer’s, and proposed that revving up protective signals or shutting down destructive ones could point to therapeutics.
Beth Stevens of Children’s Hospital, Boston, reported that synaptic strength may govern which synapses are picked off by microglia, and which survive. As a postdoc in Ben Barres’s lab at Stanford University in California in 2007, Stevens reported that neuronal circuitry was streamlined during development by the complement cascade—a multi-protein defense system that tags pathogens and debris for destruction. The complement protein C1q, expressed in the CNS during development, tagged the surface of synapses and triggered their pruning as well (see Stevens et al., 2007). C1q initiates the complement cascade, a proteolytic sequence of events that results in the cleavage of the complement protein C3. Products of C3 cleavage then bind to the surface of the tagged synapse and beckon microglia, which express the C3 receptor, to eat them. Since helming her own lab, Stevens has begun to uncover how this cascade results in the selection of certain synapses for destruction, while sparing others.
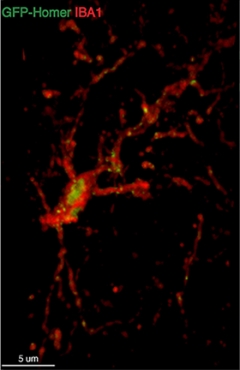
Dining on Synapses:
Microglia (red) ingest synaptic proteins (green). [Image courtesy of Soyon Hong.]
In 2012, Stevens reported that microglia preferentially target synapses that transmit the weakest signals to postsynaptic neurons. By injecting different fluorescent tracers into the eyes of mice, Stevens traced axonal projections from neurons in the retina to those in the visual cortex. When Stevens inhibited neuronal activity in one eye and not the other, she saw that microglia preferentially engulfed synapses from neurons in the blocked eye. It appeared that microglia were somehow singling out the weakest synapses for destruction (see Schafer et al., 2012). Last month in Taos, Stevens reported that this predilection for weaker synapses is dependent on C1q, suggesting that the complement cascade directs microglia to devour synapses.
To scrutinize the mechanisms behind pruning selection at the single synapse level, researchers in the Stevens lab persevered for five years to construct an in vitro neuronal circuit system. They based it on connections that form between retinal neurons that make up the optic nerve, and neurons in the dorsolateral geniculate nucleus (dLGN), a major relay station in the thalamus that receives inputs from the optic nerve and passes them onto other regions of the brain. They co-cultured two pieces of retina (each labeled with a different dye), along with a piece of embryonic dLGN from a mouse embryo. In vitro, the retinal ganglion cells sprouted axons that projected into the dLGN, where they formed synapses that underwent pruning after some time in culture.
Leaving this circuit unperturbed, axons from both slices of retina innervated most of the dLGN neurons. However, when the researchers increased synaptic transmission in one retina, synapses onto dLGN neurons from this more-active retina quickly outnumbered those from the less-active retina. Stevens said microglia in the cultures likely eliminated the weaker synapses, but only future research will confirm that. Interestingly, complement dictated this competition, because when the cultures were prepared from C1q-deficient mice, activated or inhibited retinal ganglion cells equally innervated the dLGN. Stevens said her lab will continue to use this new tool to focus in on selection at the level of individual synapses.
In addition to the destructive signal delivered by C1q, Stevens said her lab was investigating a potential protective signal that discourages microglial pruning. She plans to use the lab’s retinal culture model to determine how the two signals interact, and whether the protective signal is expressed on some synapses and not others.
Carla Shatz of Stanford University presented recent findings about PirB, another regulator of synaptic pruning. As previously covered by Alzform, Shatz reported that the major histocompatibility complex class I (MHCI) receptor is expressed on neurons, and keeps synaptic plasticity in check by promoting synaptic pruning. Mice that lack the receptor display enhanced ocular dominance, a classic example of neuronal plasticity in which neurons emanating from an open eye form more synaptic connections than those projecting from a closed one. Interestingly, Aβ activates PirB, kicking pruning into overdrive (see Sep 2013 news). Shatz proposed that blocking the receptor could be a way to prevent the synaptic destruction that occurs in the early stages of AD, even before plaques appear.
To this end, Shatz and colleagues generated a soluble version of PirB to act as a decoy to distract Aβ and other ligands and block activation of PirB signaling on neurons. Injecting the decoy directly into mouse brains not only drove ocular dominance plasticity during development (when the process normally occurs), but in adult mice as well. When the researchers covered a single eye in mice for their first six weeks of life, normal mice were left permanently blinded in that eye, whereas PirB-deficient mice gradually regained much of their vision (see Bochner et al., 2014). Shatz plans to test whether blocking PirB in early stages of AD could fend off cognitive decline associated with synaptic destruction. Prompted by a question from the audience, Shatz said that the PirB-deficient mice outperform normal mice on nearly all cognitive tasks tested so far.
Marco Colonna of Washington University School of Medicine in St. Louis was intrigued by the expression of PirB on neurons, and asked if it was the only member of the large Pir protein family to be expressed on those cells. Colonna was among the first researchers to discover the human homologue of PirB, Lilrb2 (also known as ILT4), and had reported the receptor’s expression on peripheral myeloid cells (see Colonna et al., 1998). Shatz said that PirB is the only Pir protein detected on neurons, and added that according to the Allen Brain Atlas, Lilrb2 is expressed in the human brain.
From Development to Disease
Could overzealous synaptic disposal contribute to AD? Soyon Hong of the Stevens lab addressed that question in her Keystone talk. Because evidence points to synaptic loss due to Aβ oligomers as an early step in AD pathogenesis, Hong set out to measure whether the complement-mediated pruning that Stevens documented during development revs up again at the earliest stages of disease (see Selkoe, 2002; and Koffie et al., 2009). She looked for signs of complement-mediated synaptic pruning in the J20 model of AD. Hong found that a breakdown in synaptic connectivity in vulnerable regions of the brain preceded the appearance of plaques. This synaptic disconnection depended upon the complement cascade. The same process occurred in normal mice injected with Aβ oligomers, and correlated with microglial activation and their engulfment of some synapses. This dovetails with previous findings from both Stevens’ lab and Cynthia Lemere’s lab at Brigham and Women’s Hospital, Boston. It indicated that C3 deficiency preserved synapses and cognition in AD mice, even though these mice had a heavier plaque burden than their C3-expressing brethren (see Aug 2013 news).
Hong proposed that C1q latches onto neurons and triggers the complement cascade. This would result in the tagging of those neuronal synapses with C3, which triggers microglia expressing the C3 receptor to engulf synapses. Hong said she is investigating the relationship between Aβ oligomers and C1q binding to neurons.
Researchers at the meeting were impressed by the findings about synaptic pruning. “Both Shatz and Stevens’ data are exciting,” said Hugh Perry of the University of Southampton in England. “A few years ago, I was skeptical about the idea of this selective synaptic pruning, and wondered how it would work. Now, it seems there is a cohesive story coming together.” Perry added that more work remains to be done to ascertain whether selective pruning plays a role in human disease.
Shatz and Stevens plan to investigate how the mixed messages they identified—i.e., PirB, complement, and protective signals—blend together to target specific synapses for destruction, both in development and disease. Shatz told the audience that she hopes the research will one day lead to treatments that promote synaptic plasticity in aging people at a level closer to that achieved in the young. She added, jokingly, “I’m getting older and I want the pill.”—Jessica Shugart
References
News Citations
- Immune Receptor Binds Aβ Oligomers, Spurs Synaptic Loss
- Spine Shrinkers: Aβ Oligomers Caught in the Act
- Curbing Innate Immunity Boosts Synapses, Cognition
Research Models Citations
Paper Citations
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007 Dec 14;131(6):1164-78. PubMed.
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012 May 24;74(4):691-705. PubMed.
- Bochner DN, Sapp RW, Adelson JD, Zhang S, Lee H, Djurisic M, Syken J, Dan Y, Shatz CJ. Blocking PirB up-regulates spines and functional synapses to unlock visual cortical plasticity and facilitate recovery from amblyopia. Sci Transl Med. 2014 Oct 15;6(258):258ra140. PubMed.
- Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O'Callaghan CA, Dunbar R, Ogg GS, Cerundolo V, Rolink A. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998 Apr 1;160(7):3096-100. PubMed.
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002 Oct 25;298(5594):789-91. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.