Spine Shrinkers: Aβ Oligomers Caught in the Act
Quick Links
Though it’s now widely accepted that amyloid-β oligomers are bad news for dendritic spines, most of the data supporting this contention comes from cell culture or tissue slice experiments. Showing that Aβ shrivels up spines in vivo has been a tougher nut to crack, mostly because of limitations imposed by microscopy. But in this week’s PNAS online, researchers led by Tara Spires-Jones at Massachusetts General Hospital, Charlestown, get to the kernel of the matter using array tomography, a form of serial microscopy that has the resolution needed to identify individual spines. The results confirm that in a transgenic mouse model of Alzheimer disease, spines vanish from the vicinity of plaques. Because spine density and density of oligomeric Aβ correlate exquisitely with distance from plaques, the results suggest that plaques may be a reservoir of oligomers, which leach out and promote the collapse of dendritic spines, thus “reconciling the apparently competing schools of thought of ‘plaque’ vs. ‘oligomeric Aβ’ as the synaptotoxic species in the brain of AD patients,” write the authors.
Curiously, the paper also reports that some spines are significantly smaller than others in the transgenic animals and—perhaps paradoxically—even in control mice, irrespective of how close the spines are to a plaque. What does correlate with small spine size is binding to Aβ oligomers. “That was one of the most interesting things for me that came of the study, that synapses are contacted by oligomers—even in the control animals,” Spires-Jones said in an interview with ARF. The authors found Aβ oligomers in contact with smaller spines in non-transgenic mice, which have neither human APP nor mutant presenilin and which do not form amyloid plaques. Based on this observation, the authors suggest that oligomeric Aβ might even have a normal physiological role, for example, in long-term depression (LTD), which is known to cause reduction in spine volume. “To the best of my knowledge, oligomers of Aβ have not been seen in control mice brain before,” said Spires-Jones.
This work comes courtesy of array tomography microscopy, a method pioneered by Stephen Smith at Stanford University (see ARF Bar Harbor report), and by NAB61, an Aβ oligomer-specific antibody raised by Virginia Lee’s lab at the University of Pennsylvania, Philadelphia (see Lee et al., 2006). Both Smith and Lee are coauthors on the paper. First author Robert Koffie and colleagues used array tomography to prepare ribbons of 50 nanometer-thin sections of postmortem brain tissue from APP/PS1 transgenic mice and controls. They took serial images of the ribbon and then generated a 3D image of whole stacks of sections. Spires-Jones said that she had tried for years to get good quality images of plaques and spines from postmortem tissue, but even with the best tricks up her sleeve could only get resolution of 2-3 microns, larger than the half-micron synapse. “If we saw colocalization in those thick sections we could just be seeing something above or below the synapse,” she said. But array tomography brings the resolution down to the 50 nm scale.
Using the technique, Koffie and colleagues looked at how the size and number of synaptic spines (as judged by staining for the post-synaptic marker PSD-95) correlated with plaques. They found that the spine density was much lower near plaques, but radiating outward increased in a linear fashion to control levels at about 50 μM from the plaque halo (see figure below). They also found that synapses were smaller in size (by about 40 percent) in transgenic animals compared to controls, though this reduction did not change with distance from plaques. The size reduction does, however, correlate with the presence of Aβ oligomers. Using the NAB61 antibody, Koffie and colleagues found that there is a halo of oligomeric Aβ surrounding plaques, and that the density of the oligomers falls off with increasing distance from the plaque core, just as the synaptic density does. Furthermore, they found that a subset of post-synaptic densities colocalize with Aβ oligomers and that this colocalization is much greater than would be predicted by chance alone. Finally, they showed that those post-synaptic densities in contact with Aβ oligomers were significantly smaller (~40 percent) than those that were Aβ-free. The work suggests that even far from plaques, Aβ oligomers are toxic to synapses. “This is a confirmation of oligomeric Aβ being toxic not just in tissues, cells in culture, and in mice when you inject it, but actually at the synapse in a living system,” said Spires-Jones.
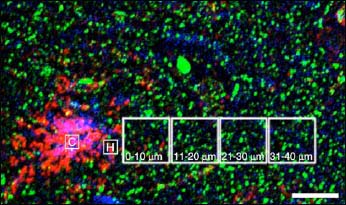
Far and Away
Dendritic spine (green) density and oligomeric Aβ burden (red) both correlate with distance from dense core Aβ plaques, visualized by thioflavin S staining (purple). Plaques may act as a resevoir of Aβ oligomers, which leach out and damage dendritic spines. Image credit: National Academy of Sciences
The work fits with the idea that Aβ oligomers can cause long-term depression (LTD) and loss of dendritic spines (see ARF related news story). The authors even suggest that because smaller dendritic spines are associated with LTD, their findings might be pointing to a physiological role for oligomeric Aβ. That oligomers in control mouse brain tissue also colocalize with smaller spines supports that idea. Though NAB61 was raised against human Aβ, the sequences may be conserved enough between mouse and human that the antibody cross-reacts with the mouse Aβ as well, and the researchers confirmed this biochemically. “It could also be that the antibody works in mouse because it is specific for the oligomeric conformation and not for monomers,” said Spires-Jones. The assumption is that despite primary sequence differences, human and mouse Aβ assume similar tertiary structures when they form oligomers.
The next step will be to look at human tissue. Spires-Jones said that she is already collecting samples for that purpose. One downside of array tomography is that tissue must be obtained freshly for embedding, which precludes material from frozen tissue banks. Spires-Jones is also planning to examine human control tissue. Since many cognitively normal people have been found to have a high plaque burden on postmortem, it may be interesting to see if the same correlations between Aβ oligomer burden and synaptic density losses are also apparent in those individuals. “There is a lot of compensation in humans. You could already have neurons around plaques shrinking but neural systems are able to compensate,” she suggested. There may also be a genetic component to the whole dynamic. “Based on your genetic makeup you may be able to clear oligomers or plaques more effectively,” she said.—Tom Fagan
References
News Citations
- Enabling Technologies for Alzheimer Disease Research: Seventh Bar Harbor Workshop, 2007, Part 4
- AMPA Receptors: Going, Going, Gone in Aβ-exposed Synapses, PSD95 Knockouts
Paper Citations
- Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006 Feb 17;281(7):4292-9. PubMed.
Further Reading
News
- Enabling Technologies for Alzheimer Disease Research: Seventh Bar Harbor Workshop, 2007, Part 4
- How Does Aβ Do Harm? New Clues on Insulin Signaling, Spines, Inflammation
- Popcorn Plaque? Alzheimer Disease Is Slow, Yet Plaque Growth Is Fast
- AMPA Receptors: Going, Going, Gone in Aβ-exposed Synapses, PSD95 Knockouts
Primary Papers
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):4012-7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Florence
This is a very important paper illustrating for the first time at high resolution the relation between Abeta oligomers and the condition of dendritic spines in a highly significant animal model of AD. Obviously, several points remain to be addressed, for instance the presence of AMPA and NMDA receptors in the neurite membranes immediately surrounded by the oligomers (they could reveal a distribution similar to that imaged for the dendritic spines with respect to Abeta oligomer gradient) and the levels of free calcium in neurons contacted by Abeta oligomers. However I trust that, when provided, those results will confirm the direct effect of Abeta oligomers on the neuritic membrane.
Another point that must still be clarified is the following: if Abeta oligomers leak from mature fibrils found in the plaques, why in many cases do people bearing plaques not suffer the symptoms of AD? I think that a possible explanation can be searched in several recent papers indicating that Abeta and other proteins can polymerize into fibrils with different structural features, and hence stabilities, depending on several factors including environmental conditions. It could well be that those plaque-bearing people who do not suffer AD have highly stable Abeta fibrils that actively and unidirectionally recruit Abeta monomers and oligomers before they can damage neurons. It would be interesting if the authors check for the presence of Abeta oligomers around the plaques in samples from plaque-bearing asymptomatic people.
Banner Research Institute
The paper by Koffie et al., by showing correlation between oligomeric Aβ and PSD loss, adds significantly to our appreciation of mechanisms by which flavors of APP, especially of Aβ, attack synapses in Alzheimer disease. There are now publications that demonstrate Aβ induced decrements not only of postsynaptic sites (Koffie, et al., 2009; Lacor et al., 2004) but also of presynaptic entities (e.g., Kelly, et al., 2005; Yao et al., 2003; Callahan et al., 1999). But what is the contribution of synaptic deficits to the cognitive declines of AD? The early studies of DeKosky and Scheff (1990) and Terry et al. (1991) agree in finding a correlation of about 0.70 between postmortem measures of synapse density and antemortem scores on cognitive tests. However, a correlation of 0.7 yields an R2 of about 0.50 which leaves 50 percent of the variance in cognitive scores unaccounted for by synapse density. Where might the missing 50 percent lie? Of course, it is presumptuous to assert that synapse density in one small tissue block from a single brain region should explain a phenomenon as complex as cognition. However, let us proceed. May part of the missing 50 percent be attributed to the fact that reduced synapse density in a shrinking cortex means an even greater loss of synapses than that indicated by density data alone? May part of the missing 50 percent be attributed to data indicating that even synapses that are structurally present may not be optimally functional—as evidenced by data showing reduced expression of dynamin 1 in AD (Kelly et al., 2005; Yao et al. 2003), by reduced expression of synaptophysin by single neurons in association with tau phosphorylation or tangles (Callahan et al., 1999), as well as by reductions in transmitter systems (e.g., Lanari et al., 2006 for recent, brief review)? And, to what extent might decrements in these indices of synaptic functional capacity be a consequence of or a cause of synaptic loss?
Data from imaging studies suggest that we need to look more broadly than at the synapse alone in attempting to correlate cognitive deficits of AD to neurobiological variables. For example, such studies demonstrate the importance of metabolic function in cognitive decline (e.g., Villain et al., 2008; Alexander et al., 2002). Gene expression array studies also indicate that transcripts that are not directly synaptic play significant roles in the pathophysiology of AD. These include transcripts related to metabolism, transport, oxidative phosphorylation, protein localization, myelin and inflammation, as examples (e.g., Liang et al., 2008; Miller et al., 2008; Wilmot et al., 2008; Blalock et al., 2004; Colangelo et al., 2002).
Synapses are unquestionably central players in the pathophysiology of AD, and we are starting to understand how various flavors of Aβ, as well as other molecules, act on synapses in AD. But it is clear that there are also other important players. How they relate to each other, which are drivers and which are driven; where they all fit into the pathophysiological cascade of Alzheimer’s disease remains to be established.
References:
Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer's Disease Treatment Studies. Am J Psychiatry. 2002 May;159(5):738-45. PubMed.
Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004 Feb 17;101(7):2173-8. PubMed.
Callahan LM, Vaules WA, Coleman PD. Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J Neuropathol Exp Neurol. 1999 Mar;58(3):275-87. PubMed.
Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002 Nov 1;70(3):462-73. PubMed.
Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990 May;27(5):457-64. PubMed.
Kelly BL, Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006 Sep 22;281(38):28079-89. PubMed.
Kelly BL, Vassar R, Ferreira A. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem. 2005 Sep 9;280(36):31746-53. PubMed.
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):4012-7. PubMed.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004 Nov 10;24(45):10191-200. PubMed.
Lanari A, Amenta F, Silvestrelli G, Tomassoni D, Parnetti L. Neurotransmitter deficits in behavioural and psychological symptoms of Alzheimer's disease. Mech Ageing Dev. 2006 Feb;127(2):158-65. PubMed.
Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA. Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4441-6. Epub 2008 Mar 10 PubMed.
Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008 Feb 6;28(6):1410-20. PubMed.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991 Oct;30(4):572-80. PubMed.
Villain N, Desgranges B, Viader F, De la Sayette V, Mézenge F, Landeau B, Baron JC, Eustache F, Chételat G. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J Neurosci. 2008 Jun 11;28(24):6174-81. PubMed.
Wilmot B, McWeeney SK, Nixon RR, Montine TJ, Laut J, Harrington CA, Kaye JA, Kramer PL. Translational gene mapping of cognitive decline. Neurobiol Aging. 2008 Apr;29(4):524-41. PubMed.
Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, Coleman PD. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis. 2003 Mar;12(2):97-109. PubMed.
Harvard Medical School
This article by Koffie et al. contributes importantly to elucidating the contribution of amyloid plaque pathology to synapse loss in Alzheimer’s disease. Heretofore, studies examining the effects of Aβ on synapse morphology have been performed primarily in ex vivo paradigms; however, this work sheds light on spine dynamics at the plaque interface in vivo.
Decreased synapse density has been well documented in human brain affected by AD (1). Importantly, the extent of synapse loss correlates with the severity of dementia, a finding also applicable to individuals with mild cognitive impairment (2, 3). Aβ is most commonly implicated as the pathogenic species responsible for the initial insidious loss of synapse density (4-6). While biochemical and genetic evidence suggests that accumulation of parenchymal Aβ is a critical initiator, a finding requiring reconciliation is that amyloid plaque burden does not correlate strongly with the severity of disease (7,8). Soluble Aβ, on the other hand, correlates strongly with disease severity, and specifically oligomeric assembly forms are the ones to demonstrate robust effects on synapse physiology. It is within this context that Koffie et al. examines how Aβ oligomers loosely associated at the periphery of neuritic plaques affects synapse density. While prior work by Spires-Jones et al. (9) demonstrated that synapse loss was most pronounced within 30 μm of plaques, the identity of the toxic species contained within this complex β-sheet rich structure remained unidentified. By using array tomography and an antibody characterized as Aβ oligomer-specific (10), the authors here present compelling evidence that implicates oligomeric Aβ surrounding insoluble plaques as the likely culprit inducing synapse loss.
The authors first demonstrate using in vivo multiphoton imaging and postmortem sectioning that the vast majority of compact plaques contain a penumbra that is positive for Aβ oligomers as judged by NAB61 immunostaining. Array tomography is subsequently used to illustrate that synapse density is reduced within the compact portion of plaques as well as in the oligomer-rich halo region. The magnitude of synapse loss is blunted with increasing distance, with this significant difference extending up to 20 μm radially from the halo margins. The authors also describe that the NAB61 signal co-localizes with PSD-95 puncta, suggesting an interaction of Aβ oligomers with post-synaptic components. Of physiologic relevance, spines associated with Aβ oligomers were smaller, suggestive of a morphologic manifestation of synapse depression. Taken together, these findings are all highly suggestive that soluble Aβ oligomers are enriched at amyloid plaques and can ultimately initiate the synapse loss observed in AD brain.
This powerful combination of in vivo multiphoton imaging, postmortem analysis with array tomography, and Aβ oligomer-specific reagents opens up a new avenue for understanding AD pathophysiology. With these tools in hand and other ones now emerging, the field is now poised to visualize answers that have been elusive in the past. For instance, it may be of interest to address whether spine density is similarly affected in regions surrounding diffuse plaques that are concentrated in Aβ oligomers. Given that compact plaques contain a panoply of non-Aβ factors, the relatively more simple composition of diffuse plaques may be able to provide additional direct evidence that Aβ oligomers are sufficient to induce synapse loss. Second, is this synapse loss reversible? Given the recent clinical trials examining the therapeutic advantage of passive immunization, NAB61 (or other oligomer-specific antibodies) may provide insight as to whether clearance of soluble higher order Aβ assembly forms can prevent or reverse the dramatic spine loss surrounding amyloid plaques. Third, application of more recently developed higher resolution imaging techniques, such as stimulation emission depletion (STED) microscopy, may provide clearer visualization of the more subtle changes in synapse structure that take place near plaques as described in this study or elsewhere in the parenchyma. Lastly, the authors suggest a physiologic role for Aβ oligomers in regulating synapse function. A cross of APP transgenic mice with channel rhodopsin-2 (ChR2), halorhodopsin (NpHR) mice (11) may be useful to further examine the general role of network dysfunction (12,13) in Aβ oligomer rich parenchyma, such as the penumbra surrounding compact plaques described by Koffie et al. More specifically, by expressing these optically activated ChR2/NpHR ion channels, neuronal activity can be focally modulated revealing perturbations in the circuitry surrounding amyloid plaques.
References:
Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987 Apr;78(2):151-64. PubMed.
Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001 Jan 9;56(1):127-9. PubMed.
Scheff SW, Price DA, Schmitt FA, Dekosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007 May 1;68(18):1501-8. PubMed.
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006 Dec 7;52(5):831-43. PubMed.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007 Jan 24;27(4):796-807. PubMed.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007 Mar 14;27(11):2866-75. PubMed.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991 Oct;30(4):572-80. PubMed.
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999 Dec;46(6):860-6. PubMed.
Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005 Aug 3;25(31):7278-87. PubMed.
Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006 Feb 17;281(7):4292-9. PubMed.
Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature. 2007 Apr 5;446(7136):633-9. PubMed.
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007 Sep 6;55(5):697-711. PubMed.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
University of California San Diego
This paper confirms, in vivo, a role for soluble Aβ oligomers in the disassembly of synapses surrounding plaques. The authors for the first time apply array tomography to quantitatively assess the interaction between postsynaptic densities/spines with microdeposits of oligomeric Aβ present in a halo extending from the edge of the dense core of plaques. Interestingly, they find that the reduction in the density but not in the size of postsynaptic densities is inversely correlated to the distance from the plaques. Overall, this paper suggests that in vivo plaques act as a source of toxic soluble oligomeric Aβ, which directly interacts with dendritic spines, causing their disappearance. However, these data don’t explain why 60 percent of postsynaptic densities and dendritic spines resist the toxic effects of Aβ, or why plaques in elderly individuals are not always associated with cognitive decline. Maybe the answer for the latter point can be found in a recent paper (Lesne et al., 2008) where the authors studied plaque-bearing mice with reduced levels of oligomeric Aβ assemblies and find that they have intact memory function. Finally Koffie et al. describe oligomeric Aβ puncta either juxtaposed to PSD clusters or on the extracellular surface of dendritic spines (see Fig. 4C) and occasionally of presynaptic compartments. However, due to limitation in their technique they cannot exclude the possibility that the Aβ puncta, even though they appear on the extracellular surface of dendritic spines, could be in reality on or within the glia processes surrounding individual synapses.
References:
Lesné S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. 2008 Feb 6;151(3):745-9. PubMed.
University of Toronto
Several papers in 2009 have explored the physiological consequences of Aβ oligomers in Alzheimer disease. In this study from Brad Hyman’s lab, a novel method enabling precise quantification of small structures was adopted to study the presence of Aβ oligomers in Alzheimer brains. The technique, based on immunofluorescence on ultrathin tissue sections, is called array tomography. The lab group found that oligomeric Aβ is deposited as a halo around senile plaques in the Alzheimer brain, but that virtually no oligomers could be found more distant than 50 μm from the plaques. In a second part of this work, transgenic mouse brains were analyzed. Here, micro-deposits of oligomeric Aβ were found to be associated with a subset of excitatory synapses. Interestingly, those synapses were considerably smaller than synapses not in contact with oligomeric Aβ. This work adds to our knowledge about both the relationship between plaques/oligomers and about the pathogenic role of Aβ oligomers in the affected brain.
View all comments by Martin IngelssonMake a Comment
To make a comment you must login or register.