All Roads Lead to TREM2: Gearing Up to Target This Receptor
Quick Links
Since the discovery of rare, yet potent risk variants in the TREM2 gene 11 years ago, the microglial receptor has emerged as a pivot point in the pathogenesis of Alzheimer’s disease. Findings presented at the International Conference on Alzheimer’s and Parkinson’s Diseases, held March 28 to April 1 in Gothenburg, Sweden, fleshed out TREM2 signaling mechanisms that could be amenable for therapeutic targeting.
- Deleting iRhom2 shifts TREM2 from shedding to signaling.
- TGF-β2 receptor placed into TREM2 cleavage cascade by CSF GWAS.
- Small molecule agonists trigger TREM2.
- CSF chemokines cast as biomarkers of TREM2 activation.
More AD risk genes were implicated in the TREM2 signaling pathway. The protein encoded by the risk gene rhomboid family member 2 apparently stabilizes a protease that snips TREM2 off the microglial surface. Levels of this soluble piece (sTREM2) in the cerebrospinal fluid served as the basis for an entire GWAS, from which more genes, including one encoding TGFβ receptor 2, were found to tweak TREM2 cleavage. Some scientists showcased preclinical findings on up-and-coming small-molecule TREM2 agonists, while others identified chemokines as potential biomarkers of TREM2 activation for future trials.
From its perch as a transmembrane receptor on the microglial cell surface, TREM2 senses an ever-growing cast of ligands, including various lipids as well as Aβ (which, in a separate talk, was proposed to be itself lipidated most of the time). TREM2 signaling can switch microglia from homeostatic to an ever-growing number of responsive states. Besides the internal signaling cascade set off by the full-length receptor, TREM2 can also be cleaved from the cell surface by metalloproteases. The resulting soluble fragment rises in CSF in the early stages of AD, and has been found to aid in the clearance of Aβ plaques, among other functions (Jan 2016 news; Jan 2018 news; Apr 2019 news). The relative roles of the full-length versus soluble forms of TREM2 in various disease states are under intense investigation.
At AD/PD, Stefan Lichtenthaler of Ludwig Maximilians University in Munich emphasized that while metalloproteases such as ADAM10 and ADAM17 are known to shed TREM2 from the surface of myeloid cells, little is known about how that is regulated, particularly in microglia. To investigate one piece of this cleavage puzzle, Lichtenthaler dug into a previously reported genetic finding, which had tied elevated expression of the RHBDF2 gene to increased risk of AD (De Jager et al., 2014). RHBDF2 encodes inactivated Rhom2 protein. As its name suggests, iRhom2 has no catalytic activity. Instead, the protein reportedly ushers the ADAM17 protease from the endoplasmic reticulum to the trans-Golgi network, where the protease becomes activated before moving out to the plasma membrane and cleaving its substrates (McIlwain et al., 2012; Adrain et al., 2012). TNFα is one of ADAM17’s infamous substrates and, in myeloid cells lacking iRhom2, the marooning of ADAM17 thwarts the release of this potent inflammatory cytokine. Might iRhom2 have a similar effect on another ADAM17 substrate … TREM2?
Sure enough, Georg Jocher and colleagues from Lichtenthaler’s lab found that, in microglial cell lines and in primary microglia, knockout of iRhom2 drastically reduced the secretion of sTREM2, while bolstering expression of the full-length receptor. The resulting uptick in TREM2 signaling promoted a transcriptional shift into the disease-associated microglia (DAM) state. Notably, when Jocher unleashed microglial cells onto plaque-laden brain slices from APP/PS1 mice, he found that iRhom2 deficiency boosted their phagocytosis of plaques by 50 percent. However, removal of iRhom2 also doubled the proportion of microglia harboring lipid droplets, suggesting alterations in lipid metabolism. At the moment, it’s unclear whether these lipid droplets signify stepped-up internalization, and/or a slowdown in endolysosomal processing in the iRhom2-deficient microglia. Lichtenthaler later noted that in contrast to the consequences of iRhom2 deficiency, treatment with TREM2 agonist antibodies reduces these droplets.

iRhom2, the Handler. The iRhom2 proteins ushers ADAM17 from the ER up to the plasma membrane, where it can cleave TREM2. iRhom2 deficiency inhibits sTREM2 cleavage, promoting signaling through the full-length receptor.
The findings cast iRhom2 as a new genetic modifier of sTREM2 release, and confirmed ADAM17 as a major TREM2 protease in microglia, Lichtenthaler concluded. He proposed that in addition to agonistic TREM2 antibodies, perhaps blocking iRhom2 could be a way to enhance signaling through the full-length receptor.
Inhibiting TREM2's cleavage may enhance signaling through the full-length receptor, but it also halts production of soluble TREM2, which itself could also affect microglial function, noted Beth Stevens of Boston's Children’s Hospital. How do the new data address this dichotomy? Lichtenthaler acknowledged the issue. He said that in addition to reducing soluble TREM2, iRhom2 deficiency also puts the kibosh on release of TNF-α and CSF-1R. “A goal is to understand whether blocking iRhom2 is purely beneficial in the context of AD, or may also have unexpected detrimental consequences, potentially due to the reduced cleavage of other ADAM17 substrates,” Lichtenthaler wrote to Alzforum.
Bart De Strooper of UK Dementia Research Institute in London saw a silver lining in these potentially multipronged effects, noting that iRhom2 inhibition could theoretically hit two birds with one stone—strengthening TREM2 signaling while also quelling neuroinflammation wrought by TNF-α. Lichtenthaler agreed, noting that SciRhom, a Munich-based biotech company he is unaffiliated with, is developing small-molecule iRhom2 inhibitors for the treatment of inflammatory disorders involving TNF-α. He added that because iRhom2 is a multipass transmembrane protein that travels to the plasma membrane, it is particularly amenable to targeting with antibodies. However, further investigation is needed to fully grasp its function within the brain. To that end, Lichtenthaler’s group is using iRhom2-deficient mouse models of amyloidosis. Thus far, these mice suggest that halving iRhom2 assuages amyloidosis.
Approaching the investigation of TREM2 mechanisms from another angle, Carlos Cruchaga of Washington University in St. Louis and colleagues paired genomics and proteomics to hunt for factors involved in TREM2 signaling and shedding. To do this, they compiled CSF samples from more than 3,000 participants across eight AD research cohorts, measured thousands of proteins—soluble TREM2 among them—and ran a GWAS to find variants associated with its concentration. Four loci reached genome-wide significance.
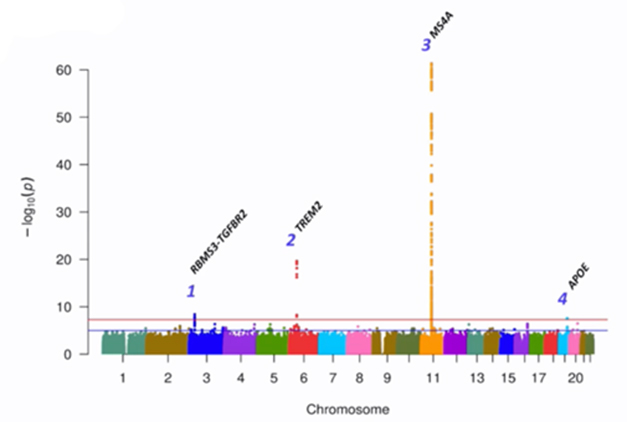
Ties to sTREM2. Across the genome, four loci significantly associated with the CSF concentration of sTREM2. [Courtesy of Carlos Cruchaga, Washington University.]
One was TREM2 itself. Specifically, carriers of the R47H AD risk variant tended to have less sTREM2 in their CSF than noncarriers. Another signal came from MS4A. This AD risk locus previously has been implicated in tweaking TREM2 cleavage and, more recently, in microglial lipid metabolism (Aug 2019 news; Mar 2023 news). In the CSF GWAS, two variants within this multigene locus—one in the MS4a4a gene and the other in the MS4a6a gene—associated with higher sTREM2 levels.
A third GWAS signal popped up in the ApoE locus. Surprisingly to Cruchaga, the association was wholly independent of ApoE2, E3, or E4 genotype. What could it be? In other datasets, this polymorphism associated with high mRNA expression of a nearby gene, Nectin-2, and its encoded protein, poliovirus receptor-related 2 (PVRL2), Cruchaga told Alzforum. A transmembrane protein with Ig-like domains, PVRL2 stands accused of rolling out the welcome mat for herpesviruses, which themselves have been implicated in AD. Nectin-2 variants have also been tied to AD risk, but the gene’s close proximity to ApoE has complicated efforts to study its independent relationship with disease (Zhou et al., 2019). The findings hint that PVRL2 could play a bona fide role in in AD risk by influencing TREM2 homeostasis.
The fourth hit came from a locus harboring two genes—RBMS3 and TGFBR2. Further experiments in cultured macrophages suggest that the latter, which encodes the TGFβ2 receptor, was the one influencing CSF sTREM2, Cruchaga reported. While he does not know how this variant shifts sTREM2 levels, he did note that signaling through the TGFβ2 receptor is known to influence microglial responses, and the findings cast this receptor as yet another therapeutic target. Dovetailing with that idea, Oleg Butovsky of Brigham and Women’s Hospital, Boston, reported at AD/PD that ApoE4 promotes TGF-β signaling in microglia, locking the cells in a nonresponsive, homeostatic state. Butovsky, too, suggested TGF-β signaling as a therapeutic target.
Finally, Cruchaga told the audience that among the more than 3,000 participants included in the analysis, higher CSF sTREM2 correlated with a reduced risk of AD. This jibes with previous data from the DIAN cohort, where mutation carriers with the strongest uptick in CSF sTREM2 had a slower worsening of disease (Mar 2022 news).
In toto, what do the findings say about full-length versus soluble TREM2 signaling in AD? Cruchaga hesitated to suggest that one is more important than the other. “Obviously full-length TREM2 is a signal transduction protein, but at the same time, we think soluble TREM2 is doing something that may be protective,” he said. “We need more studies about the human biology of TREM2.” How to target Trem2, then? Cruchaga favors the idea of promoting TREM2 signaling with agonists, rather than solely blocking its cleavage.
A handful of TREM2 agonists are in clinical development. Antibodies that tickle TREM2 signaling, including Denali’s DNL919 and Alector’s AL002 are being evaluated in Phase 1 and 2 trials, respectively, though neither reported results at AD/PD. Vigil Neuroscience, a company in Watertown, Massachusetts, is evaluating its TREM2 agonist antibody, VGL101, in people with ALSP, a rare genetic disorder caused by a mutation in the CSF-1R gene. However, for AD—a much bigger indication that will require chronic dosing—Vigil is taking a small-molecule approach. The company originally bought its TREM2 agonist antibody and a suite of TREM2 small-molecule agonists from Amgen in 2020, after that company pulled the plug on its neuroscience programs.
At AD/PD, Vigil’s Christian Mirescu showed preclinical data on the company’s lead TREM2 agonists, which have been selected and polished for brain permeability, oral availability, and potency. Mirescu claimed they work like “molecular glue,” i.e., by promoting the clustering of TREM2 receptors on the microglial cell surface. In cultured human microglia, this huddling boosted TREM2 signaling and reduced shedding. The agonists worked with the known TREM2 AD risk variants and in conjunction with endogenous TREM2 ligands. For example, one agonist upped the maximal response to sulfatide, a damage-associated TREM2 ligand, sixfold.
In humanized TREM2 mice fed with a Vigil TREM2 agonist, the molecule readily crossed into the brain. There, it incited microglia to shift into a disease-associated state, and stoked brain levels of IFN-γ inducible protein (IP-10), a chemokine that recruits other immune cells. These responses resembled those triggered by injection with a TREM2 agonist antibody, Mirescu reported. Feeding the small molecule agonist to cynomolgus macaques triggered a dramatic drop in levels of soluble TREM2 in their CSF over the first 24 hours, followed by a return to baseline levels three days later.
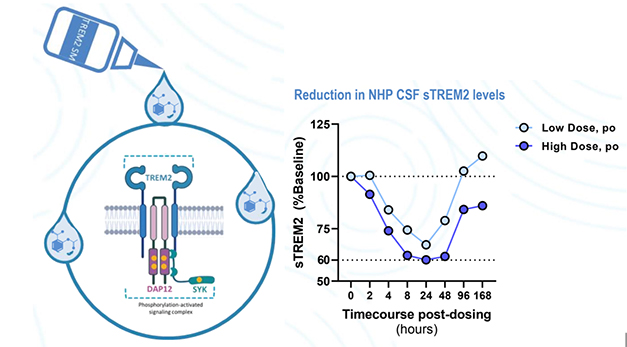
Glue That TREM2. Vigil’s small-molecule TREM2 agonists promote clustering and signaling through the full-length receptor (left). They prevent TREM2 cleavage and shedding, as seen by a drop in CSF sTREM2 in monkeys fed a single dose (right). [Courtesy of Christian Mirescu, Vigil Neuroscience.]
Vigil aims to file an IND application for its lead TREM2 agonist in the second half of 2023. Before testing the agonists in the broader AD population, the company will evaluate them in people who carry loss-of-function mutations in TREM2 or other microglial genes, Mirescu told Alzforum.
Even as the field gears up for trials, it still lacks specific ways to track TREM2 signaling. Sure, soluble TREM2 levels may dip in the CSF following engagement of some agonists, but how will scientists know that their agonists are enhancing signaling through the receptor? To that end, Choya Yoon of Merck shared results from her lab's hunt for such TREM2 activation biomarkers.
Yoon surveyed a panel of cytokines and chemokines in brain homogenates from the TgCRND8 mouse model of amyloidosis, on a wild-type or TREM2-deficient background. She found that two chemokines—Ccl4 and IP-10—ramped up in response to amyloidosis, but only when mice expressed TREM2. Yoon also found Ccl4 and IP-10 to be elevated in the mouse CSF, but not plasma, in response to amyloidosis. Both cytokines were detectable in the CSF of cognitively normal people, suggesting they could be feasibly tracked in trials, Yoon said.
How might these chemokines respond to TREM2 agonists? Yoon tested this in different mouse models. In TgCRND8 mice, both chemokines spiked in the CSF 24 hours after treatment with an agonistic, anti-mouse TREM2 antibody. This also happened in humanized TREM2 mice treated with an anti-human TREM2 antibody developed by Merck.
Curiously, neither IP-10 nor Ccl4 budged after repeated dosing, perhaps reflecting a desensitization of the chemokine response after chronic TREM2 stimulation, Yoon said. In ongoing experiments, she is trying to define this refractory period, in hopes of devising a proper dosing interval for clinical trials. The Merck scientists are also measuring these chemokines in CSF from people across the AD spectrum, and are correlating them with the other known AD biomarkers. Yoon's biomarker talk at AD/PD reflected part of a larger effort at Merck to understand and target Trem2.—Jessica Shugart
References
News Citations
- TREM2 Goes Up in Spinal Fluid in Early Alzheimer’s
- New Mouse Models Reveal Unexpected Property of TREM2
- Cut Loose, Soluble TREM2 Beckons Microglia to Mop Up Plaques
- Paper Alert: MS4A Variants May Sway Alzheimer’s Risk Via TREM2
- Alzheimer’s Gene MS4A4A Governs the State of Microglia
- Robust TREM2 Expression May Delay Alzheimer’s Disease
Mutations Citations
Therapeutics Citations
Research Models Citations
Paper Citations
- De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, Tang A, Raj T, Replogle J, Brodeur W, Gabriel S, Chai HS, Younkin C, Younkin SG, Zou F, Szyf M, Epstein CB, Schneider JA, Bernstein BE, Meissner A, Ertekin-Taner N, Chibnik LB, Kellis M, Mill J, Bennett DA. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014 Sep;17(9):1156-63. Epub 2014 Aug 17 PubMed.
- McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC, Lang KS, Häussinger D, Wakeham A, Itie-Youten A, Khokha R, Ohashi PS, Blobel CP, Mak TW. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. 2012 Jan 13;335(6065):229-32. PubMed.
- Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science. 2012 Jan 13;335(6065):225-8. PubMed.
- Zhou X, Chen Y, Mok KY, Kwok TC, Mok VC, Guo Q, Ip FC, Chen Y, Mullapudi N, Alzheimer’s Disease Neuroimaging Initiative, Giusti-Rodríguez P, Sullivan PF, Hardy J, Fu AK, Li Y, Ip NY. Non-coding variability at the APOE locus contributes to the Alzheimer's risk. Nat Commun. 2019 Jul 25;10(1):3310. PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.