Preclinical Research Offers New Angles on Immunotherapy
Quick Links
Despite early disappointments, drug developers have bought into immunotherapy for Alzheimer’s disease, banking on a handful of antibodies and active vaccines currently in Phase 2/3 trials and others in Phase 1. As evident from this year’s Society for Neuroscience meeting, held October 17-21 in Chicago, the trend continues. Researchers are still developing immunotherapies for Aβ and for tau, building in new models and strategies to study how comorbidities, such as vascular disease, factor in. Researchers in Italy have even used intrabodies—antibodies produced inside cells—to study the intracellular dynamics of Aβ oligomerization.
Immunotherapy ‘Passifies’ Pyroglutamate Aβ
Most immunotherapies against Aβ have been designed with full-length Aβ1-42 in mind, but some groups have been targeting the truncated Aβ3-x form that undergoes glutamic acid cyclization at the N-terminal. Helen Crehan, who works in Cynthia Lemere’s lab at Brigham and Women’ Hospital, Boston, reported that antibodies to the pyroglutamate (pGlu) form were effective in a treatment paradigm but only if they have good effector function.
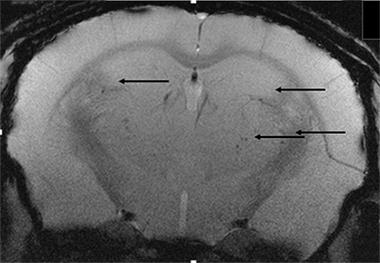
Complicating Co-morbidities.
Microhemorrhages (arrows) increase when mice with vascular disease are treated with Aβ antibodies. [Image courtesy of Donna Wilcock.]
Lemere recently published findings that these antibodies can prevent plaques and protect against behavioral deficits when given prophylactically to young APPSwe/PS1ΔE9 mice (Frost et al., 2015). In Chicago, Crehan reported that when given to 12-month-old male mice, which have rampant amyloid plaques, weekly injections of a mouse IgG2a isoform reduced pGluAβ immunoreactivity in the hippocampus at 16 months. Total Aβ and Aβ plaque burden fell as well, indicating that the therapy reduced more than just pGluAβ. “It could be that pGluAβ acts as a seed and we prevented deposition because we removed that seed,” said Lemere. As an alternative, she suggested that microglia activated by the antibody could remove all forms of Aβ. Interestingly, Crehan detected no significant reduction in plaque load when she treated mice with a mouse IgG1 variant of the same antibody. Mouse IgG1 antibodies do not bind Fc receptors on immune cells and therefore have less effector function than their IgG2a homologs. In keeping with the idea that good effector function is necessary for these antibodies to clear Aβ, Crehan reported last March at the International Conference on Alzheimer's and Parkinson’s Diseases in Nice, France, that in ex vivo phagocytosis assays microglia mop up more Aβ when treated with the IgG2a antibody than with any other isoform.
The IgG2a antibody also improved learning and memory. In the water T maze, treated APP/PS1 mice outperformed controls after three months of therapy, though they performed less well than wild-type mice, said Lemere. Nevertheless, Probiodrug, a German company targeting pGluAβ, plans to begin clinical trials of a humanized version of the mouse IgG2a antibody to pGluAβ. Phase 1 trials are slated to begin in 2017.
Active Vaccine for Tau
In a different take on viral delivery, researchers for Kiran Bhaskar’s lab at the University of New Mexico, Albuquerque, described a virus-like particle vaccine targeting pathological phospho-tau. By using chemical cross-linkers, researchers can load empty viral capsids with antigens, and the particle then elicits a strong immune response from B cells. Others have used the strategy for vaccines against Aβ, including Novartis’s CAD-106, which is in Phase 2/3. Bhaskar’s group has used the same Qβ particle to present phospho-tau peptides to the immune system. In her talk, Nikki Maphis said that she can pack about 180 peptides (TPPAPKpTPPSSGQG) from the proline-rich domain of tau onto one virus-like particle.
How well does this work as a vaccine? Maphis tested it in rTg4510 mice, which overexpress human tau with the P301L mutation. They have phosphorylated tau and memory problems at three months and neurofibrillary tangles by about four months of age. Beginning when they were two months old, Maphis gave 10 mice three biweekly injections of p-Tau-VLP or a control particle. After six weeks the mice had generated a high titer of anti phospho-tau antibodies. Maphis detected the antibodies in cortical extracts.
Animals sacrificed at four months old had one-third as much p-tau in their hippocampi as controls and a similar reduction in neurofibrillary tangles in their cortices. The amount of total insoluble tau plummeted in the brain as well. The treated mice mustered fewer CD45+ microglia, indicating less inflammation, and fewer neurons appeared to be apoptotic, suggesting they were protected. The antigen also improved mouse learning and memory. The vaccinated animals did better in a novel-object-recognition test than controls treated with viral particle alone. In the Morris water maze, vaccinated mice behaved much like wild-type, spending about the same amount of time in the target quadrant in a probe test.
Other researchers wondered if the antibodies generated by the vaccine bound tau inside cells, and if so, how much of the antibody actually got to its target. The mechanism is still unclear, but Maphis said that these antibodies could be getting into cells or may be soaking up tau released into the parenchyma, possibly through synapses. Scientists debate whether and how tau spreads transynaptically (see Apr 2015 conference news). Bhaskar told Alzforum that he is looking for partners to help develop this vaccine for the clinic.
Intrabodies for the Endoplasmic Reticulum
In another twist on Aβ immunotherapy, Giovanni Meli from the European Brain Research Institute, Rome, is using intrabodies to prevent cellular damage by oligomeric forms of Aβ. Intrabodies are short, usually single-chain, antibodies that are smuggled into cells under the veil of viral or other vectors (see Jan 2002 news). Meli and colleagues led by Antonino Cattaneo at EBRI have gone a step further, using protein engineering to target the intrabodies to specific subcellular compartments (see Meli et al., 2014). The researchers added a KDEL amino acid motif to the scFvA13 intrabody, which recognizes only oligomeric forms of Aβ in a sequence- and conformation-selective manner. This motif targets proteins to the endoplasmic reticulum. Indeed, Meli found that 7PA2 cells, which express human APP with the V717F mutation, retained scFvA13-KDEL in their ER. 7PA2 cells produce large quantities of Aβ42 and pump various forms of the peptide, including oligomers, out of the cell (see Clearly et al., 2005). Cells expressing the ER-targeted antibody produced 50 to 70 percent less oligomers than did 7PA2 controls, and secreted more monomers into the medium. The research suggested that oligomers can form in the ER and that intrabodies curtail them.
In Chicago, Meli reported that this ER intrabody protects cells from Aβ toxicity. It restored the mitochondrial membrane potential in 7PA2 cells, which supports the idea that AD pathology compromises the integrity of these organelles. ScFv13 also normalized proliferation of neural stem cells from Tg2576 mice when delivered into the cells via lentiviruses. Meli said this intrabody may be useful not only to study the intracellular dynamics of Aβ oligomerization, but also as a potential therapy. He has started in vivo experiments to target the intrabody to the hippocampus of AD mouse models.
Other researchers at the meeting were intrigued by the data but wondered if the intrabody might retain Aβ in the ER, in effect forcing it to oligomerize there. Meli thought this unlikely because he detects no Aβ increase in cells treated with the intrabody.
Passive Immunotherapy and Vascular Dementia
With many vaccination strategies for Aβ and tau in development, do some work better than others for certain patients? Erica Weekman, from Donna Wilcock’s lab at the University of Kentucky, Lexington, addressed this in a mouse study of mixed vascular and parenchymal pathology. A large overlap exists between AD and vascular dementia, such that about 40 percent of AD patients have both morbidities. “Targeting an AD pathological process may be insufficient for clinical outcomes if there are also vascular contributions to the cognitive impairment,” said Wilcock.
To test this in a model, Wilcock’s group fed nine-month-old APP/PS1 mice a diet deficient in folate and vitamins B6 and B12. This drives up homocysteine, causing hyperhomocysteinemia (HHcy), a risk factor for vascular damage. This diet causes vascular cognitive impairment in wild-type mice (see Sudduth et al., 2013). After six months on this diet, the APP/PS1 animals had more microhemorrhages (see image above) and a striking redistribution of Aβ from the parenchyma to the vasculature, though the total Aβ remained the same, said Weekman. The HHcy mice made twice as many errors in a radial arm maze as did the APP/PS1 controls or HHcy alone (Sudduth et al., 2014).
Would this comorbidity affect immunotherapy? Weekman fed nine-month-old APP/PS1 mice the HHcy diet, and three months in, she treated them with weekly intraperitoneal injections of a control IgG or the 3D6 monoclonal antibody—the mouse forerunner of bapineuzumab. The monoclonal elicited a much greater reduction in Aβ in the cortices of mice on the control diet than in mice with vascular pathology. Curiously, 3D6 suppressed microglial activation in HHcy animals, whereas it activated microglia in mice on normal chow. The Aβ antibody held inflammation at bay in the HHcy mice as well, as judged by transcripts for inflammatory markers. While all this data suggested that the antibody might work in animals with mixed pathology, Wilcock believes that the immunotherapy, on top of the amyloid pathology and vascular pathology, may have tipped the innate immune system into senescence. “That’s speculative, but we are looking into it,” she said.
Further analysis indicated all was not well. APP/PS1 mice with hyperhomocysteinemia made more errors in a radial arm maze if they were treated with the antibody, and mouse MRI indicated that the antibody induced even more microhemorrhaging than in untreated controls.
Weekman concluded that anti-Aβ treatment might be unsuitable for people with vascular pathology as a comorbidity to their AD. In fact, ARIA, or Aβ-related imaging abnormalities, a sign of vasogenic edema, contributed to the demise of the bapineuzumab program. To keep edema at bay, dosing in many patients had to be reduced to levels where the antibody was ineffective (see Aug 2012 news). “We still believe that antibody binding to amyloid in the vasculature and the ensuing inflammatory response is the cause of ARIA, but this is still a hypothesis,” said Wilcock. “My guess is plasma HHcy may be a risk factor for ARIA, but that’s pure speculation at this point.”
Wilcock said that it would be valuable to test other antibodies in this mixed-pathology model. She is planning to examine correlations among HHcy, inflammation, AD pathology, and small-vessel disease to get a better handle on how these interact in people.—Tom Fagan
References
Research Models Citations
Therapeutics Citations
News Citations
- Protein Propagation Real, but Mechanisms Hazy
- Protein-Protein Interactions, Cytoskeleton Implicated in Huntington's
- Clinical Trials of Intravenous Bapineuzumab Halted
Paper Citations
- Frost JL, Liu B, Rahfeld JU, Kleinschmidt M, O'Nuallain B, Le KX, Lues I, Caldarone BJ, Schilling S, Demuth HU, Lemere CA. An anti-pyroglutamate-3 Aβ vaccine reduces plaques and improves cognition in APPswe/PS1ΔE9 mice. Neurobiol Aging. 2015 Dec;36(12):3187-99. Epub 2015 Aug 31 PubMed.
- Meli G, Lecci A, Manca A, Krako N, Albertini V, Benussi L, Ghidoni R, Cattaneo A. Conformational targeting of intracellular Aβ oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat Commun. 2014 May 27;5:3867. PubMed.
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005 Jan;8(1):79-84. PubMed.
- Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab. 2013 May;33(5):708-15. Epub 2013 Jan 30 PubMed.
- Sudduth TL, Weekman EM, Brothers HM, Braun K, Wilcock DM. β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther. 2014;6(3):32. Epub 2014 Jun 9 PubMed.
Other Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.