Protein Propagation Real, but Mechanisms Hazy
Quick Links
The once-outlandish idea that misfolded proteins can propagate through the brain along anatomical connections has gone mainstream. At the 12th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 18-22 in Nice, France, no fewer than 30 talks and 50 posters addressed the topic of protein spread, with most scientists saying there is no doubt this occurs. Beyond that bit of consensus, however, little else is clear. Researchers debated which forms of different proteins constitute the pathological players, where in the body the process starts, and exactly how these species travel through the brain and pass between cells. Whether propagating proteins should be dubbed bona fide prions or are merely “prion-like” has become a semantic point of contention.
“It’s a sticking point for many in the field, because prions were originally defined as infectious particles. I think this nomenclature issue has impeded acceptance of the concept,” Lary Walker at Emory University, Atlanta, told Alzforum.
In the last few years, numerous animal studies have shown the spread of misfolded, aggregated forms of tau, Aβ, and α-synuclein to connected brain regions (e.g., Jun 2009 news; Jul 2009 news; Nov 2012 news). In many of these experiments, the seeds came from patients’ brains, emphasizing the capacity of neurodegenerative aggregates to corrupt native protein as prions do. Postmortem and PET imaging studies suggest the same propagation process occurs in human brain (for review, see Brettschneider et al., 2015). Other studies have turned up evidence in human brain of distinct strains of tau and Aβ—another prion-like feature (see Aug 2013 conference news; Sep 2013 news).
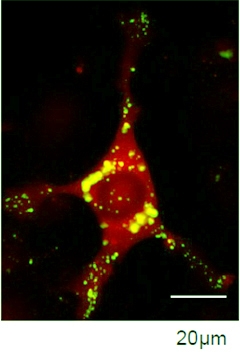
Seeds of Destruction.
Exogenous α-synuclein (green) seeds aggregation of endogenous protein (red), forming large deposits (overlay appears gold). [Image courtesy of Ronald Melki.]
What Seeds Disease?
But what exactly are these seeds, and how do they propagate? In Nice, Paul Fraser of the University of Toronto recapped the two classic models for prion-like transmission: template-directed refolding, and seeded nucleation (see Aguzzi and Calella, 2009). In one, a single misfolded protein molecule acts as the infectious agent and directs the misfolding of normal proteins one by one. The other model posits that a certain number of misfolded molecules always exist in equilibrium with healthy protein, but are harmless by themselves. It is only when a number of misfolded proteins assemble into an ordered structure that disease begins. These aggregates then recruit additional misfolded monomers, forming large protein deposits. Because the initial seeds form slowly, there is a long lag before disease begins, whereas subsequent recruitment occurs rapidly, causing pathology to run rampant through the brain. Claudio Soto of the University of Texas, Houston, believes this latter model has become accepted.
Seeds, then, are larger than monomers, but researchers disagree on how much larger. Are they oligomers? Fibrils? What initiates pathology? In the past decade, oligomers have drawn attention as a likely toxic entity, but Charles Duyckaerts of Pitié-Salpêtrière Hospital, Paris, described his recent study in which Aβ oligomers injected into mouse brains diffused widely within 24 hours (in press at Neurobiology of Aging). If soluble oligomers seed plaques, the plaques should then appear throughout the brain rather than only in regions connected to the injection site, he pointed out. He added that injected oligomers are quickly cleared by microglia, leaving no lasting damage. Soto disagreed with Duyckaerts’ interpretation that oligomers therefore do not seed disease, noting that injected prion proteins also disappear for a time before disease crops up.
To many researchers, the likely villains are fibrils. In cell culture studies, 4 nM of yeast prion fibrils are more toxic than 1 μM of oligomers, said Ronald Melki of the French National Center for Scientific Research, Gif–sur–Yvette, France (see Bucciantini et al., 2012). Patrik Brundin, of the Van Andel Institute in Grand Rapids, Michigan, noted that after injection into the olfactory bulb, pure preparations of monomers and oligomers travel efficiently between neurons but get swept up by microglia, whereas longer fibrils tend to get stuck outside cells; thus, neither form seeds well. In mouse experiments from Virginia Lee and John Trojanowski at the University of Pennsylvania Perelman School of Medicine, Philadelphia, sonicating α-synuclein fibrils to produce a mix of long and short strands produced the most efficient seeding, Brundin said. After a lag period of one to three months, this material triggered α-synuclein deposits in brain regions two synapses away from the injection site, as well as behavioral deficits (see Luk et al., 2012). “Fibrils are the species that trigger long-term pathology, but how you treat the fibril affects the dynamics of incubation time and efficiency,” Brundin concluded.
Evidence for the existence of different fibril strains is growing. Fraser showed that fibrils can have distinct tertiary structures, for example straight or twisted, each with its own stability and incubation time. Melki characterized synthetic α-synuclein aggregated under two different conditions, finding that each aggregate behaved differently in terms of its toxicity and ability to seed (see Bousset et al., 2013). Each strain has a “molecular bar code,” Melki said, jokingly comparing their shapes to different pasta, such as spaghetti versus linguini.
Posttranslational modifications may help determine how proteins aggregate into strains. Dietmar Thal of Ulm University, Germany, noted that phosphorylation and acetylation of Aβ and tau boost the number of intermediates and fibrils. The next question is to find out what forms lurk in the brains of patients. “We need to recover material from human brain and amplify it in vitro,” Melki said.
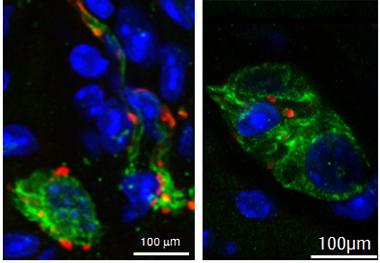
Does Parkinson’s Begin in Gut?
Deposits of α-synuclein (red) accumulate in enteric neurons (green), but not other cell types (nuclei in blue), in the gut of young transgenic α-synuclein mice. [Image courtesy of Markus Britschgi.]
Can aggregates seed disease from outside the brain? Some researchers have proposed that Parkinson’s disease could be triggered by α-synuclein accumulation in the gut, which then travels to the brainstem and midbrain (see Jul 2011 news series), or in the nose. At AD/PD, Markus Britschgi of Roche presented new data supporting the hypothesis that certain intestinal inflammations could kick off the disease (for a review, see Lema Tomé et al., 2013). Britschgi induced colitis in transgenic mice that expressed mutant human α-synuclein by putting dextran sodium sulfate in their drinking water for five days. In that time, intracellular aggregates of α-synuclein in enteric neurons roughly doubled, Britschgi said. To study longer-term effects, he induced chronic colitis in the mice for four weeks, then examined enteric neurons two months later. Although the colitis had long since resolved, the α-synuclein deposits persisted.
Many of these deposits were in macrophages. More gut α-synuclein accumulated in mice that lacked fractalkine receptor CX3CR1. This signaling pathway is known to be important for proper microglia-neuron interaction in the brain, and may likewise facilitate communication between macrophages and neurons in the gut, Britschgi said (see Nov 2011 news; Cardona et al., 2006). When these immune cells become activated by pathogens, environmental factors, or persistent deposits, they may damage gut tissue and further stress neurons, he suggested. Next Britschgi will investigate whether α-synuclein deposits eventually turn up in the brains of treated mice. Similar processes may occur in people, Britschgi noted. In eight out of 11 people with inflammatory bowel disease, Britschgi found α-synuclein deposits in gut macrophages and neurites. On the flip side, at least one study shows that people with Parkinson’s often suffer from inflammation of the colon (see Devos et al., 2013). A major genetic risk factor for PD, LRRK2, also predisposes to inflammatory bowel disease. If intestinal inflammation does precede Parkinson’s in some patients, it might provide an opportunity for early intervention, perhaps with antibacterial or anti-inflammatory agents, or even anti-α-synuclein antibodies, Britschgi wrote to Alzforum.
Some audience members countered that α-synuclein accumulation occurs in the guts of healthy controls, too, and that epidemiological studies to date have not reported a link between intestinal disease and Parkinson’s. Britschgi noted that this idea has been little studied, saying it would be worth looking for a connection in large epidemiology datasets.
Mechanisms Mostly Murky
Perhaps the greatest unknown about protein propagation is how it happens. First, here is what’s clear: Researchers agree that protein aggregates travel along anatomical networks, spreading to connecting rather than just neighboring regions. Both aggregated Aβ and tau progress through the brain in an anterograde course, or down axons, said Thal. The initial site of seeding determines which neuroanatomical pathways and networks are affected, speakers concurred. For example, tau injections in the hippocampus travel to the piriform cortex, while cortical injections sprout deposits in the hippocampus, amygdala, and midbrain, reported Diederik Moechars of Janssen Pharmaceutica, Beerse, Belgium (see Peeraer et al., 2015). Seeding of different regions determines the behavioral outcomes as well, and might be responsible for heterogeneous symptoms seen in patients, noted Ilse Dewachter of the Université Catholique de Louvain, Brussels, Belgium.

α-Synuclein Spread.
Aggregates of α-synuclein (green) spread into the anterior olfactory nucleus (pictured, nuclei in blue) after injection of fibrils in olfactory bulb. [Image courtesy of Nolwen Rey.]
To illustrate that anatomical connections are vulnerable to spread, Duyckaerts described a case of a woman who died at 85 from Alzheimer’s. A small piece of her cortex had been disconnected from the rest for 20 years, when a brain surgery severed it. In contrast to the rest of her brain, this isolated region contained no neuritic plaques or neurofibrillary tangles, only diffuse Aβ deposits. This finding highlights that the spread of pathology requires connection, Duyckaerts said.
Beyond this broad picture, however, details are fuzzy. “There is very little mechanistic data about how the process works,” Marc Diamond at the University of Texas Southwestern Medical Center, Dallas, wrote to Alzforum. How are aggregates transported inside neurons? In cell culture studies, researchers have tracked aggregates moving both anterogradely and retrogradely along axons via fast transport, in other words, carried by molecular motors along microtubules, Melki said (see Freundt et al., 2012). However, Thal noted that this remains to be proven in human brain. Aggregates might instead move along the outside of axons by guided diffusion or glial transport, he suggested.
Even less clear is how aggregates might enter and exit cells. First, about getting in. In 2013, Diamond reported that neurons take up tau and α-synuclein aggregates through a form of endocytosis, after tau sticks to heparan sulfate proteoglycans on the cell surface (see Mar 2013 conference news). To date, no replication studies have been published, nor are there papers proposing a detailed entry mechanism. At AD/PD, researchers led by Pamela McLean of the Mayo Clinic in Jacksonville, Florida, reported on a poster that α-synuclein oligomers inside exosomes were taken up equally well by cells that lacked HSPGs and cells that had them, suggesting that these molecules are not always necessary for endocytosis. Free α-synuclein, on the other hand, remained stuck to the outside of cells. Another poster, from Eliezer Masliah and colleagues at the University of California, San Diego, reported the opposite. In their hands, internalization of both oligomeric and fibrillar α-synuclein required the presence of functioning HSPGs on the cell surface. The reason for the discrepancy is not yet clear. Complicating matters, many researchers suggest that different types of aggregate may use distinct methods to infiltrate cells.
How do seeds get out? Researchers believe that neurons spit out toxic aggregates packaged in small membraneous vesicles called exosomes. Existing data indicate that only a small fraction of protein secretion occurs in these structures, but exosomes may still be crucial to the process, Brundin said. He found that lipids in these vesicles accelerated aggregation of α-synuclein fibrils in vitro (see Grey et al., 2015). Many researchers are currently probing the role of exosomes in neurodegenerative disease (see Dec 2014 conference news). Two posters at AD/PD reported evidence for transfer of Aβ aggregates inside exosomes. In one, researchers led by Efrat Levy at the Nathan S. Kline Institute for Psychiatric Research, Orangeburg, New York, described worse amyloid pathology in young Tg2576 mice injected with exosomes from older Tg2576 animals, but not in young Tg2576 mice that received exosomes from aged control mice. In the other, Martin Hallbeck and colleagues at Linköping University, Sweden, tracked the fate of fluorescently labeled Aβ oligomers in a neuronal culture system. They found Aβ inside secreted exosomes, which was taken up by other neurons through endocytosis.
Exosomes are not the only vessels in which seeding aggregates might escape cells. There are also ectosomes—larger vesicles than exosomes that get pinched off directly from the plasma membrane. They are plausible vehicles for tau, a cytosolic protein not associated with the secretory pathway, said Morvane Colin at AD/PD. Colin collaborates with Luc Buee at University of Lille, France, on mechanisms of tau spread. Colin last year had reported physiological release of tau ectosomes from cultured cells and detected them in the brain interstitial fluid of rats (Dujardin et al., 2014). At AD/PD, Colin expanded on that work. She reported that she found increased levels of tau ectosomes in the brain interstitial fluid of a new transgenic rat model of tauopathy, as well as in a hippocampal injection macaque model of tauopathy. However, because most tau is in free form, it is unclear whether these vesicular forms are the pathogenic species, Colin told the audience. A poster from Jürgen Götz and colleagues at the University of Queensland, Australia, reported that tau aggregates in ectosomes are differently phosphorylated than those in exosomes, hinting that there could be a difference in toxicity between these species.
What’s in a Name: The Prion Debate
What to call propagating proteins has become a hot-button issue. Prion researchers point out that Tau, Aβ, and α-synuclein aggregates pass most prion identity tests. Rattling off a list of characteristics, Soto noted that even some features he initially doubted have proven to exist.
For example, exposing animals to neurodegenerative seeds from various peripheral routes can stimulate brain deposition. Presenting data on peripherally administered Aβ, Rodrigo Morales in Soto’s group reported that eye drops were most efficient, followed by intraperitoneal, then intramuscular injection. Only eating Aβ aggregates failed to produce brain pathology. Morales reported that diluting Aβ aggregates delayed the appearance of pathology in linear fashion, as is the case with prion protein. The minimum amount of inoculum that produced disease was about 343 femtograms, similar to the minimum infectious dose of aggressive prion strains, he said. Mathias Jucker at the German Center for Neurodegenerative Diseases in Tübingen, Germany, previously reported that Aβ amounts as low as one femtogram could seed plaques (see Sep 2014 news).
The one test Aβ aggregates fail is that of person-to-person transmission under natural conditions, Soto said. The prion disease Creutzfeldt-Jakob also does not transmit between people through normal daily contact. Few studies have tried to measure human transmission of neurodegenerative disease, though a 2013 paper from Trojanowski and Lee reported that people who received human growth hormone from cadaver preparations were no more likely to develop Alzheimer’s or Parkinson’s than the general population (see Feb 2013 news). Researchers note that this study is not definitive, saying additional research should determine whether neurodegenerative diseases can be transferred through medical procedures under any conditions.
In fact, many researchers avoid the term “prion” to avoid stirring up confusion and fear (see Jun 2012 news). In a new review, Walker argues that redefining the term prion as a “proteinaceous nucleating particle,” rather than a “proteinaceous infectious particle,” would solve the issue. This would allow the term to encompass both prion protein diseases and neurodegenerative protein aggregates, Walker said (see Walker and Jucker, 2015). “As concepts evolve, definitions should evolve,” he told Alzforum.—Madolyn Bowman Rogers
References
News Citations
- Traveling Tau—A New Paradigm for Tau- and Other Proteinopathies?
- Aβ the Bad Apple? Seeding and Propagating Amyloidosis
- Toxic Synuclein Corrupts Native in Wild-Type Mice
- Are Protein Strains The Cause of Different Tauopathies?
- Does Aβ Come In Strains? Glimpse Into Human Brain Suggests Yes
- The Good, the Bad, and the Fractious? Ligand’s Role in Brain
- Tau, α-Synuclein Spread: Crazy Stuff—How Might It Work?
- Exosomes: Purveyors of Neurodegenerative Disease?
- Bad Seeds—Potent Aβ Peptides Instigate Plaques, Won’t Be Fixed
- In Case You Wondered: Neurodegenerative Diseases Are Not Contagious
- Aβ Sufficient for Seeding—But Is It a Prion?
Series Citations
Paper Citations
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015 Feb;16(2):109-20. Epub 2015 Jan 15 PubMed.
- Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009 Oct;89(4):1105-52. PubMed.
- Bucciantini M, Nosi D, Forzan M, Russo E, Calamai M, Pieri L, Formigli L, Quercioli F, Soria S, Pavone F, Savistchenko J, Melki R, Stefani M. Toxic effects of amyloid fibrils on cell membranes: the importance of ganglioside GM1. FASEB J. 2012 Feb;26(2):818-31. PubMed.
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012 May 7;209(5):975-86. PubMed.
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. PubMed.
- Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and α-Synuclein's Prion-like Behavior in Parkinson's Disease-Is There a Link?. Mol Neurobiol. 2012 Apr 29; PubMed.
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006 Jul;9(7):917-24. PubMed.
- Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, Neunlist M, Derkinderen P. Colonic inflammation in Parkinson's disease. Neurobiol Dis. 2012 Sep 24;50C:42-48. PubMed.
- Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E, Van Acker N, Kemp JA, Mercken M, Brunden KR, Trojanowski JQ, Dewachter I, Lee VM, Moechars D. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015 Jan;73:83-95. Epub 2014 Sep 16 PubMed.
- Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol. 2012 Oct;72(4):517-24. PubMed.
- Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, Linse S. Acceleration of α-synuclein aggregation by exosomes. J Biol Chem. 2015 Jan 30;290(5):2969-82. Epub 2014 Nov 25 PubMed.
- Dujardin S, Bégard S, Caillierez R, Lachaud C, Delattre L, Carrier S, Loyens A, Galas MC, Bousset L, Melki R, Aurégan G, Hantraye P, Brouillet E, Buée L, Colin M. Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One. 2014;9(6):e100760. Epub 2014 Jun 27 PubMed.
- Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015 Jul 8;38:87-103. Epub 2015 Mar 30 PubMed.
Further Reading
News
- Synthetic Synuclein Corrupts Native Along Mouse Brain Networks
- Insidious Spread of Aβ: More Support for Synaptic Transmission
- More Evidence Ties Aβ Strains to Distinct Pathologies
- Do Tau "Prions" Lead the Way From Concussions to Progression?
- Tales of Traveling Tau: Is Transfer Between Neurons Normal?
- Like Prions, Tau Strains Are True to Form
- An Extra Strain on the Brain—α-Synuclein Seeds Tau Aggregation
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.