Protein Liquid-Liquid Phase Transitions: The Science Is About to Gel
Quick Links
The list of mammalian proteins capable of forming liquid droplets is growing by leaps and bounds. What does this say about human biology, and what does it mean for neurodegenerative diseases, which have been tied to this phenomenon? At Phase Transitions in Biology and Disease, a meeting held May 2-3 in Leuven, Belgium, researchers grappled with these questions. What is the nature of protein droplets? Why and how do they form? What biological or pathological roles do they play, and will insight into these ephemeral organelles lead to new therapeutics? Co-organized by Joost Schymkowitz, Frederic Rousseau, Ludo Van Den Bosch, and Peter Tompa, all from VIB Leuven, the meeting drew about 150 scientists, including leading experts in the field who showed off complex biophysical theory and exquisite biology. The energy at the meeting was palpable, as was a sense that researchers are beginning to understand how liquid-liquid phase transitions fit into the biological seascape.
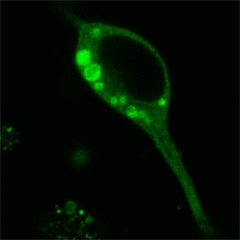
Et Tu, Tau?
GFP labeled tau forms liquid droplets in a cultured neuron. [Image courtesy of Susanne Wegmann.]
“I’m afraid we have focused too much on protein secondary structures that appear to be important in biology, but have ignored low-complexity domains,” Van Den Bosch told Alzforum. Those low-complexity domains—also called prion-like domains—are what drive proteins into liquid phase structures. “We don’t know how such proteins come together to form stress granules, the nucleolus, and other membraneless structures, but it’s a major challenge to find out,” he said.
If this meeting had a take-home message, it was that researchers are making headway. This progress is coming none too soon, according to Paul Taylor from St. Jude Children’s Research Hospital, Memphis, Tennessee. “It has become evident that phase transition is a major underlying principle of cellular organization,” he told Alzforum. “There are discrete membraneless organelles such as the nucleoli, RNA speckles, P granules, etc., but on a more transitory and smaller scale, assemblies such as transcription factories, DNA repair machines, clusters of membrane receptors, mitotic spindle assemblies, are all controlled by phase transitions,” he noted.
Researchers know that proteins linked to neurodegenerative diseases, such as FUS, TDP-43, hnRNPA1, and other RNA-binding proteins, are capable of undergoing phase separation, and this might increase their odds of eventually forming irreversible and toxic aggregates. In Leuven, they debuted new proteins that contribute to liquid droplets. Why, hello, tau! Others debated the disassembly of liquid granules—how it happens and why it matters. One question is whether specific interactions play any role in forming liquid organelles, and, if so, what those might be. Finally, though it’s early days, researchers fantasized about ways of controlling phase separation as a means to protect against toxicity. Read on for highlights of the meeting.
Turbid Tau
We can now add tau to the list of proteins that assume liquid form. Susanne Wegmann, from Bradley Hyman’s lab at Massachusetts General Hospital, Charlestown, reported that the microtubule binding protein undergoes liquid-liquid phase transitions to form droplets both in vitro and in vivo. The phase separation depended on phosphorylation; what’s more, phospho-tau droplets rapidly hardened into hydrogels that seeded tau aggregation.
Like low-complexity domains, tau is mostly intrinsically disordered and is highly charged. This, Wegmann and colleagues believe, might be enough to cause it to condense into droplets, even though it lacks true low-complexity domains found on FUS, TDP43, and hundreds of other proteins. Working with Anthony Hyman (no relation to Brad), at Germany’s Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Wegmann set about to test this idea by using crowding agents. Those are chemicals, such as polyethylene glycol (PEG) and Ficoll polysaccharides, that reduce the amount of water available to solutes and thus mimic to some extent the densely packed nature of the cellular cytoplasm.

P-tau Droplets.
Phosphorylated tau phase-separates to form liquid droplets in vitro. [Image courtesy Susanne Wegmann.]
Wegmann showed that at concentrations as low as 1.5 μM, the same range as in neurons, tau formed liquid droplets in vitro in the presence of crowding agents. These drops were dynamic, fusing together readily and recovering rapidly after photobleaching. This, in turn, indicates a rapid exchange with free tau outside the droplet. Fusion and rapid FRAP (fluorescent recovery after photobleaching) are considered hallmarks of bona fide liquid protein droplets. Tau droplets also formed in primary neurons and in living mouse cerebral cortex, said Wegmann.
Are these liquid forms of tau physiological? Wegmann hypothesized that the droplets normally provide a ready supply of highly concentrated tau that the cell calls upon to rapidly stabilize microtubules. In fact, a paper from Anthony Hyman’s group bears this out. Co-authored by Wegmann and posted to the Cold Spring Harbor Laboratory preprint server, bioRΧiv, it reports that tubulin partitioned into these droplets of liquid tau nucleates the formation of microtubule bundles (see Hernandez-Vega et al., 2017).
What about liquid tau in pathogenesis? Wegmann showed slides of brain tissue taken from a Braak stage III AD patient. Neurons without neurofibrillary tangles but that had accumulated phosphorylated tau appeared to have droplets of tau in the cytosol. The finding hinted that these drops might be a site for early tau aggregation, said Wegmann. Droplets are proposed sites for aggregation of other fibrillary proteins, such as TDP-43 and FUS (Oct 2015 webinar). In keeping with this idea, Wegmann reported that hexane1,6-diol, which destabilizes β-sheet structures, blocked tau liquid-liquid phase transitions. Aggregates that formed on the surface of tau droplets in vitro bound thioflavin S, indicative of β-sheet structure. Wegmann said these aggregates get so hard that, after a day, they hold up to atomic-force microscopy at high imaging forces. Interestingly, when she tested highly phosphorylated forms of tau, they formed droplets more rapidly than less-phosphorylated tau and formed protein aggregates more readily as well. Her findings are in keeping with hyperphosphorylated tau’s propensity to aggregate.
Researchers at this conference were not surprised that tau formed liquid droplets; however, they questioned some of the biophysical tests, particularly those suggesting that β-sheet structures were germane to the liquid-liquid phase transition. Some pointed out that the hexane-diol may not be break up β-sheets as well as claimed, and that it might have other effects. Researchers also noted that thioflavin S emits photons when it cannot dissipate energy through rotation or vibration, and that this might occur as readily in a densely crowded droplet as when bound to a β-sheet structure. Wegmann acknowledged that there is more work to do, but noted that the increase in thioflavin S fluorescence was likely too great to be explained by rotational or vibration effects.
Wegmann’s findings complement previous work from Benjamin Wolozin’s lab at Boston University. Previously, Wolozin reported that the RNA-binding protein TIA1, a major component of RNA stress granules, regulated tau behavior and toxicity. By trapping tau in stress granules, TIA1 promotes tau misfolding and aggregation, Wolozin proposed (May 2016 news). In keeping with this idea, Wegmann reported that RNA promotes tau droplets. “Among all the proteins that have been linked to FTD/ALS, tau has always been a puzzle, since it is not an RNA-binding protein, but now the pieces are falling into place,” said Wolozin. He also presented new mouse data that show how knocking down TIA1 can rescue behavior in a mouse model of tauopathy (see below).
Disassembly Required
In his talk, Taylor strengthened the connection between TIA1 and pathology. In collaboration with Ian Mackenzie at the University of British Columbia, Vancouver, and Rosa Rademakers at the Mayo Clinic, Jacksonville, Florida, he has found mutations in TIA1 that cause ALS. Those mutations are all in the low-complexity domain of the protein, and they shift the dynamic of phase transition to favor formation of protein aggregates. They seem to do this by stabilizing the protein in the liquid state, increasing the chances for nucleation of solid protein aggregates. Taylor reported that under heat stress, cells expressing TIA1 mutants form RNA granules as usual, but when the crisis passes those granules stick around instead of dissipating as they do in cells expressing normal TIA1. “In TIA1 mutants, granule formation is normal, but disassembly is protracted and incomplete,” said Taylor. He suggested this phenomenon may be common to mutations of TIA1, FUS, TDP43, hnRNPA1, and other RNA-binding proteins that undergo liquid-liquid phase separation. In effect, a failure to dissipate makes it more likely that proteins in those membraneless organelles will aggregate into solid fibrillar structures, he said.
Taylor noted that the function of another ALS gene, valosin-containing protein (VCP), fits with this concept. He and others had previously reported ALS-linked mutations in VCP (Dec 2010 news), a segregase that extracts ubiquitin-tagged proteins from large complexes. In Leuven, Taylor described how dynamic exchange of TIA1 and other proteins in and out of liquid organelles slows down if VCP is missing or blocked, and that stress granules then fail to dissipate normally.
All this hinges on ubiquitination of proteins in the organelle, Taylor proposed. Stress granule disassembly requires an E3 ubiquitin ligase, such as the TRIM21 and TRIM25 proteins that emerged from a VCP interactome analysis, as well as VCP adaptor proteins, such as NPL4, and UFD1L. These four proteins are recruited to stress granules. In the absence of any one of them, stress granules don’t properly disassemble, reported Taylor. Incidentally, Trim21 has been linked to clearance of tau aggregates (Jan 2017 news). All told, a picture began to emerge that disassembly of the membraneless organelles may be crucial for cells to stay healthy.
The True Function of Prions?
Simon Alberti, Max Planck Institute of Molecular Cell Biology and Genetics, built on the disassembly theme in his talk. Alberti studies liquid-liquid phase transitions of FUS (Sep 2015 news). He showed stunning images of solid aggregates growing out of droplets of liquid FUS, much like Taylor and others had shown for droplets of heteronuclear RNA-binding proteins (Oct 2015 news). But it was Alberti’s data on the yeast protein Sup35 that got the most attention in Leuven. In short, he posited that the role of the low-complexity, prion-like domain of this protein is to form reversible gels that protect the protein from damage.
This yeast prion forms amyloids, but Alberti showed that when budding yeast are starved, Sup35 forms transient structures. These disappear once the cells are fed again, implying they are not fibrillar. Instead, the transient structures fuse together and recover quickly after photobleaching, suggesting they are liquid droplets. However, over time, those droplets become more stable. Alberti showed cryo-EM images revealing that the Sup35 had transmogrified into an elaborate mesh-like structure.
What controls the Sup35 phase transition? Alberti reported that a physiological drop in pH that occurs under starvation drives the liquid-liquid phase separation of Sup35. He was able to induce the transition, and reverse it, by lowering and raising the pH, respectively. Even the mesh-like structure dissipated when the milieu was returned to neutral pH. Alberti reported that the NM domain in the middle portion of Sup35, which is packed with charged amino acids, is responsible for sensing the pH and driving phase separation. Remarkably, this property seems conserved in fission yeast, which separated from budding yeast hundreds of millions of years ago.
When Alberti removed the low-complexity NM domain, the C terminal by itself formed large aggregates, but they didn’t readily disassemble. Furthermore, wild-type yeast grew rapidly when they emerged from starvation, whereas yeast lacking the NM domain of Sup35 accumulated protein particles and remained dormant. “This suggests to us that the NM domain is critical for surviving stress,” said Alberti. “It changes Sup35 phase behavior from irreversible to reversible so that the cell is able to reuse the protein.” In Alberti’s model, Sup35 assists with protein translation under normal conditions, and in times of stress forms gels that easily can be reversed when the cells begin translating once more. “The low-complexity prion domain may be an evolutionary invention to protect the C terminal from aggregation under times of duress,” said Alberti. “In short, the real function of the prion domain may be to modify the phase behavior of this stress-sensitive protein in cells,” he said.
Alberti’s talk drew ample questions. People wanted to know more about the mesh structure, particularly its size and how it differs from the structure in droplets. Others wanted to know how the C terminal self-interacts and how pH governs that. Wegmann wondered if, in effect, the low-complexity domain acts like an internal chaperone, preventing the C terminal domain from aggregating. “Exactly,” said Alberti. “We think many of these [low-complexity] domains are turning proteins that are potentially dangerous into something more benign and retrievable after stress.”
Roy Parker, University of Colorado, Boulder, echoed this idea when he reported that plant late embryogenesis abundant (LEA) proteins can replace prion-like or low-complexity domains in animals. Plants make LEA proteins to protect against extreme drought. By forming generic interactions with macromolecules, they keep them from denaturing when water has evaporated. Swapping LEA low-complexity domains for those in Dhh1, a component of RNA P granules, enabled yeast to form and disassemble granules normally. That suggests that low-complexity domains, or intrinsically disordered regions, as they are also called, can behave generically.
That said, Parker does not believe that RNA granules form willy-nilly. “In vitro, many proteins can phase separate if coaxed by crowding agents, for example, but in cells the process seems much more controlled,” he said. No more so than in the case of P granules, which release RNAs in synapses and play a role in synaptic plasticity (Kiebler and Bassell, 2006). “Where is the specificity for such entities?” Parker asked. The take-home message, he said, was that RNA granules arise through both specific interactions among RNAs and RNA-binding proteins, and promiscuous liaisons among proteins with low complexity regions.
Parker proposed that the latter may be there to control the RNA-RNA interactions, which would otherwise run amok. He showed how RNA homopolymers polyU, polyC, and to a lesser extent polyA readily liquid phase separate (polyG forms quadruplex structures that precipitate), as does total yeast RNA. He noted that the concentration of RNA in a cell during a stress response can be as high as 900 μg/ml. “That is incredibly high. At that concentration, we should expect RNA molecules to form complexes with themselves and with other RNAs, and expect that to contribute to granule assembly,” he said. Parker thinks that specific interactions drive assembly of certain RNA granules, while other granules may be an unavoidable consequence of having high concentrations of RNA in a crowded cellular environment. “Cells might actually have to work to avoid them from forming,” he said.
Taylor finds this intriguing. “That RNA-RNA interactions are important to the assembly of RNA granules was fairly well established, but that the raison d’etre for RNA granules is to control RNA-RNA interactions is a novel twist,” he said. “That’s quite an influential concept.”
Tweaking the System
Though liquid-liquid phase separation is an emerging field, scientists in Leuven were already optimistic that it could lead to new strategies to tackle disease. “I absolutely believe that this field is ripe for new therapies,” said Taylor. He noted that companies have made substantial investments in this regard and that he fields constant inquiries from venture capitalists about manipulating phase transitions as a means to modulate disease.
As a proof of principle, Lindsay Becker, who works at Aaron Gitler’s lab at Stanford University, outlined how knocking down ataxin-2 expression improved symptoms in a mouse model of ALS. Becker and colleagues had previously found that the RNA-binding protein Pbp1, a yeast homolog of ataxin-2, makes TDP43 more toxic. Given that ataxin-2 mutations increase the risk for motor neuron disease, the researchers suspected that ataxin-2 did the same to TDP43 in neurons. Becker recently reported in Nature that crossing TDP43-overexpressing mice with Ataxin-2-negative mice yielded offspring with better motor control and longer lives than their ataxin-2-positive siblings. Antisense oligonucleotides that knock down ataxin-2 achieved similar results (Apr 2017 news). The knockdown seems to work because without ataxin-2, less TDP43 phase-separates into stress granules.
Similarly, Wolozin showed in his presentation that reducing TIA1 appeared to work in a mouse model of tauopathy. Researchers in his lab crossed PS19 mice, which express the P301S mutant tau, with TIA1 homozygous and heterozygous knockouts. Cortical synaptophysin and Nissl staining indicated that reducing TIA1 boosted the number of both synapses and neurons, and the crosses outperformed PS19 mice in Y-maze and novel-object-recognition memory tests. The crosses also live a lot longer. Wolozin said that at 15 months old, half of them are still alive, whereas PS19 mice have died by that age. Interestingly, all these improvements seem to occur in the face of more tau neurofibrillary tangles and paired-helical fragments. Wolozin thinks that TIA1, by corralling tau in stress granules, accelerates the formation of soluble oligomers of tau, which may be the toxic entity. He found much less of these species in the TIA1 knockouts. “Our findings are in in vivo proof that reducing an RNA–binding protein can dramatically inhibit disease progression in the PS19 mouse,” Wolozin said.
In his talk, James Shorter, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, presented what many said was a groundbreaking discovery that could lead to new therapies. In particular, it could provide a way to drive mis-localized proteins such as FUS and TDP43 back into the nucleus from the cytosol, where they tend to accumulate in neurodegenerative diseases.
Shorter reported that nuclear transportins help break apart aggregates of proteins with low-complexity domains. Shorter has been using the heat-shock protein Hsp104 to this end in a variety of cell studies (see, e.g., Yasuda et al., 2017; Nov 2015 conference news), but while Hsp104 is widely conserved, it has no homolog in animals. Shorter thought that some other protein must fill in. He looked to the karyopherin nuclear transporters. These receptors guide proteins with nuclear-localization signals from the cytosol into the nucleus. In effect, they temporarily chaperone the proteins so that they can squeeze through the small pores in the nuclear envelope. Shorter wondered if they might also disaggregate proteins with low-complexity domains. In fact, FUS, TDP43, hnRNA-binding proteins, and many other nuclear proteins contain the specific PY nuclear localization signal that the transportins recognize.
Shorter reported that karyopherin-β2 (Kapβ2) solubilizes fibrils of FUS, but not if FUS is missing its PY nuclear localization signals, and not if Kapβ2 is mutated to no longer recognize that signal. It also solubilized TAF15, hnRNPA2, EWSR1 and other RNA-binding proteins, but not TDP-43. Shorter showed that another karyopherin—Kapβ1—and importin-α take on that role for TDP-43. Kapβ2 also prevented FUS and other proteins from aggregating in vitro, while importin-α and Kapβ1 did the same for TDP-43. The karyopherins were also capable of dissolving hydrogels in vitro.
What about in cells? Together with Taylor’s lab, Shorter found that Kapβ2 prevents the ALS-linked R521C FUS or hnRNAP1 and 2 from being recruited into stress granules. In fruit flies, Shorter found that upregulating Kapβ2 in motor neurons mitigates their neurodegeneration in the face of mutant FUS and more than doubled their life span. In a model of muscle degeneration caused by the D290V mutation in hnRNAPA2, upregulating Kapβ2 restored the RNA-binding protein to the nucleus from the cytoplasm where it aggregates, and mitigated muscle degeneration.
In summary, Shorter said nuclear import receptors now should be considered chaperones and disaggregases. He suggested that small molecules that upregulate Kapβ2 or stabilize the interaction between it and mutant forms of FUS and other proteins linked to neurodegenerative disease might prove valuable.
Others were impressed by the data. “I think it’s a very important discovery that nuclear transportins can act as disaggregases,” said Wolozin. “For me that talk was a highlight of the meeting,” said Van Den Bosch. He thinks it epitomizes how this field is beginning to gel (pun intended). “At one time we thought we were looking at completely separate things—stress granules, phase transitions, aggregates, nucleocytoplasmic transport deficits—now we begin to see clearly how they are all connected,” he told Alzforum. “Clearly, we are far from understanding it all, but now we can link things in logical and relevant ways that we can investigate.”—Tom Fagan
References
Webinar Citations
News Citations
- Stress Granule Protein Entwines and Misfolds Tau
- Adding ALS to the Manifestations of VCP Mutations
- Antibodies Co-Opt Anti-Microbial Response to Clear Intraneuronal Tau
- ALS Protein Said to Liquefy, Then Freeze en Route to Disease
- Do Membraneless Organelles Host Fibril Nucleation?
- Two For One? ASOs for Ataxin Allay ALS and SCA2 in Mice
- Listen Up, Gene Silencing Strikes a Chord at RNA Meeting
Research Models Citations
Paper Citations
- Hernández-Vega A, Braun M, Scharrel L, Jahnel M, Wegmann S, Hyman BT, Alberti S, Diez S, Hyman AA. Local Nucleation of Microtubule Bundles through Tubulin Concentration into a Condensed Tau Phase. Cell Rep. 2017 Sep 5;20(10):2304-2312. PubMed.
- Kiebler MA, Bassell GJ. Neuronal RNA granules: movers and makers. Neuron. 2006 Sep 21;51(6):685-90. PubMed.
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol. 2017 Apr 3;216(4):1015-1034. Epub 2017 Mar 15 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Max Planck Institute for Biophysical Chemistry
Liquid-liquid phase separation of intrinsically disordered proteins has been implicated as a key process for the regulation of dynamic biological processes and has been linked to neurodegenerative diseases such as frontotemporal dementia and amyotrophic lateral sclerosis (Aguzzi and Altmeyer, 2016; Banani et al., 2017; Hyman et al., 2014). This study by Hernandez-Vega and colleagues shows that the microtubule-associated protein tau is able to phase separate in solution in the presence of chemical agents that mimic molecular crowding. These findings are in line with findings from the Han and Kosik labs, which showed that tau forms liquid droplets upon complex coacervation with RNA (Zhang et al., 2017), and our studies, in which we showed that the microtubule-binding domain of tau is able to phase separate in solution even in the absence of RNA or molecular crowding agents, a process commonly referred to as self-coacervation (Ambadipudi et al., 2017). While the Han/Kosik and our studies indicated that the strong increase in tau concentration within liquid droplets is critical for pathogenic aggregation of tau in Alzheimer’s disease, further supported by unpublished data from Susanne Wegmann and colleagues, this study by Hernandez-Vega demonstrates that tau droplets not only provides the means to strongly increase local tau concentration (i.e. tau reaches a supersaturated state [Ambadipudi et al., 2017]), but also to recruit tubulin dimers to tau droplets. The recruitment of tubulin dimers to tau droplets lifts the tubulin dimer concentration above the critical concentration for tubulin polymerization, resulting in the formation of long microtubule filaments.
A highly interesting finding is that tubulin was not only recruited to tau droplets but also stimulated tau droplet formation. This might be rationalized by the molecular properties of tubulin dimers. Both α- and β-tubulin contain negatively charged tails at their carboxyl-termini. These negatively charged tails have been suggested to be important for binding to tau (Serrano et al., 1984). The negatively charged tubulin tails might thus act similar to RNA and heparin to promote complex coacervation of the positively charged microtubule-binding domain of tau with negatively charged molecules (Ambadipudi et al., 2017; Zhang et al., 2017).
Another interesting observation is that a sole increase in tubulin concentration was not sufficient to generate microtubule bundles in a homogeneous environment, suggesting that the environment of the drop is important for generation of microtubule bundles in liquid droplets. A critical question that needs to be addressed is therefore how droplet formation changes the structural properties of tau. Indeed, my lab recently showed that short residue stretches in the microtubule-binding domain, comprising the conserved PGGG motifs, fold into a hairpin-like structure upon binding to the interface between tubulin heterodimers (Kadavath, Hofele et al., 2015; Kadavath, Jaremko et al., 2015). Droplet formation might thus favor this hairpin formation and promote cross-linking of microtubule filaments into bundles. The intrinsically disordered nature of the tau proteins, with its four microtubule-binding sites in the central repeat domain of tau (Kadavath, Hofele et al., 2015; Mukrasch et al., 2009), is expected to be critical for this process.
Overall, the three now-published studies from the Han/Kosik, Diez/Hyman, and my labs stress the importance of liquid-phase separation for the function and pathology of the tau protein. These are exciting times in tau biology.
References:
Aguzzi A, Altmeyer M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Biol. 2016 Jul;26(7):547-58. Epub 2016 Apr 1 PubMed.
Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun. 2017 Aug 17;8(1):275. PubMed.
Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017 May;18(5):285-298. Epub 2017 Feb 22 PubMed.
Hyman AA, Weber CA, Jülicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39-58. PubMed.
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A. 2015 Jun 16;112(24):7501-6. Epub 2015 Jun 1 PubMed.
Kadavath H, Jaremko M, Jaremko Ł, Biernat J, Mandelkow E, Zweckstetter M. Folding of the Tau Protein on Microtubules. Angew Chem Int Ed Engl. 2015 Aug 24;54(35):10347-51. Epub 2015 Jun 19 PubMed.
Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009 Feb 17;7(2):e34. PubMed.
Serrano L, de la Torre J, Maccioni RB, Avila J. Involvement of the carboxyl-terminal domain of tubulin in the regulation of its assembly. Proc Natl Acad Sci U S A. 1984 Oct;81(19):5989-93. PubMed.
Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, Han S. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017 Jul;15(7):e2002183. Epub 2017 Jul 6 PubMed.
Make a Comment
To make a comment you must login or register.