Virtual Workshop Tackles LATE, a Cause of Late-life Dementia
Quick Links
A virtual workshop on limbic predominant age-related TDP-43 encephalopathy, held virtually February 11, began with a moment of silence for the late John Trojanowski. His work played a critical role in nabbing TDP-43 inclusions as a culprit behind frontotemporal lobar degeneration and amyotrophic lateral sclerosis, and later, he helped peg the same RNA-binding protein as the critical component in what is now known as LATE—a vastly more common neuropathological entity that comes with memory loss.
- LATE neuropathologic change detected in 39 percent of older people at autopsy.
- More than 90 percent of cases are distinct from FTLD-TDP.
- For cases that are tough to call, more definitive exclusion criteria are needed.
At the workshop, researchers joined from across the aisles of FTD/ALS and AD research to carve out a niche for LATE, which was christened as a distinct neuropathological disease only three years ago. Since then, researchers have painstakingly plumbed brain samples from multiple autopsy cohorts, further refining the neuropathological characteristics of LATE-neuropathological change (NC), its prevalence, and how this flavor of TDP-43 pathology contributes to cognitive decline, particularly in comparison to its more famous amnestic doppelganger, AD.
As researchers at the meeting presented these findings from cohort after cohort, it became clear that LATE-NC is common, cropping up in around a third of brain samples from people who died in their 80s and 90s. Several investigators reported that this limbic pathology contributes substantially to cognitive decline, either by itself or in combination with AD, casting LATE as a public health threat in its own right. It may also be a meddler in clinical trials for AD, researchers lamented, because it can accelerate that dementia. As of now, a LATE diagnosis can only be made at autopsy, and biomarkers are sorely needed to detect it during life. A few contenders—including fluid and neuroimaging biomarkers—ended the meeting on a promising note.
Co-chaired by Peter Nelson of the University of Kentucky in Lexington and Julie Schneider of Rush University in Chicago, the five-hour meeting hosted by the NIA drew more than 350 attendees from 14 different countries. It featured 19 talks on the neuropathological, clinical, epidemiological, and genetic aspects of LATE, along with 25 prerecorded poster narrations. The workshop convened researchers from different “cliques” in the dementia field, Nelson said. This was by design. “To have all these factions talking to each other, and listening to each other, was very productive,” Nelson told Alzforum.
Others agreed. “It was important to bring different people together to really define the urgent questions that need to be resolved about LATE,” said Manuela Neumann of the German Center for Neurodegenerative Diseases in Tübingen.
“I was pretty blown away by the whole meeting,” said Robert Rissman of the University of California, San Diego, who presented preliminary findings on a new biomarker for TDP-43. “There’s a lot of excitement in this field right now, but still so much we don’t know,” Rissman said.
TDP-43 arrived on the scene in 2006, when Neumann, then a visiting scholar in Trojanowski’s lab at the University of Pennsylvania, Philadelphia, identified ubiquitinated inclusions of the protein as the hallmark pathology in many cases of FTLD and ALS (Oct 2006 news; Cairns et al., 2007). Soon after, neuropathologists started detecting TDP-43 inclusions in people with AD and/or hippocampal sclerosis, which causes severe neuronal loss and gliosis in the hippocampus (Amador-Ortiz et al., 2007). In these cases, which tended to have AD-like symptoms rather than symptoms of FTLD or ALS, TDP-43 inclusions predominated in the limbic regions, including the amygdala and hippocampus.
Not all cases of this limbic TDP-43 pathology come with hippocampal sclerosis, but vice versa, researchers have found that the vast majority of hippocampal sclerosis is accompanied by limbic TDP-43. In recognition of this pattern, a cadre of researchers held a workshop in 2018 to forge a consensus on the nature of the beast. From that meeting, “LATE” was born, along with a proposed neuropathological staging scheme (May 2019 news). Based primarily on autopsy data from the Religious Orders Study (ROS-MAP), researchers proposed that TDP-43 inclusions appear in the amygdala in stage 1, then the hippocampus and entorhinal cortex in stage 2, and finally the neocortex in stage 3 (James et al., 2016). Cognitive impairment tended to start once the pathology spread beyond the amygdala. As its name suggests, LATE arises late in life, lurking in the brains of 20 percent of people in their 80s who had received a dementia diagnosis, at least in the ROS-MAP cohort.
At the time, not everyone was jazzed about the new moniker. Some researchers questioned the need to name a new disease after what was primarily a neuropathological phenomenon with uncertain clinical effect (Josephs et al., 2019). They also called for further work to distinguish LATE-NC from FTLD.
In response to the latter challenge, Nelson led a study that tasked neuropathologists with discriminating between FTLD-TDP-43 and stage 3 cases of LATE-NC from autopsy cohorts at the University of Kentucky and the University of Pennsylvania (Robinson et al., 2020). Using prespecified criteria based on where and how severe TDP-43 pathology was in the brain—limbic areas for LATE and frontal cortex for FTD—the pathologists, who were blind to the diagnosis of the patients, correctly distinguished between the two disorders more than 90 percent of the time. When cases with less severe stages of LATE-NC were included in the analysis, overlap of regional pathology between the two diseases shrank. The specificity of telling the two apart rose to 98 percent, because TDP-43 pathology in earlier stages of LATE-NC is more tightly confined to limbic regions, clearly distinguishing it from FTLD.
Better Staging
At the meeting, Alexandra Young of King’s College London described a staging scheme based on SuStain, a machine-learning analysis that uses neuropathological data from hundreds of brain samples to identify patterns of pathology progression (Oct 2018 news). Young previously used SuStain to identify different subtypes of AD (Apr 2021 news). Applying the algorithm to brain samples from the University of Pennsylvania brain bank, Young identified stages of LATE-NC that roughly aligned with previously proposed staging schemes, with TDP-43 aggregates starting in the amygdala, moving into the hippocampus and entorhinal cortex, and finally out into the neocortical regions. The model also distinguished between LATE-NC, FTLD-TDP, and ALS patterns of neuropathology 96 percent of the time. This was regardless of whether people with LATE-NC also had AD. “That’s a better diagnostic specificity than we can achieve with many dementia pathologies,” Nelson said during his presentation at the meeting, referring to both his published findings and Young’s new data.
The difference between most cases of FTLD and LATE-NC might appear obvious to the discerning eye of a neuropathologist, or a computer algorithm; even so, the official neuropathological criteria for LATE-NC need to be updated to reflect these distinctions, Neumann emphasized in her talk at the meeting. Specifically, she noted that based on the current criteria, all cases of FTLD-TDP-43 and 30 to 60 percent of ALS cases, in which TDP-43 inclusions are found in the frontal cortex, could technically be classified as stage 3 LATE-NC.
Neumann thinks that clearly defined exclusion criteria are needed. For example, if TDP-43 inclusions crowd the primary motor cortex, as in ALS, that would eliminate LATE-NC. Similarly, if cortical inclusions are not Type A—defined as small, compact, cytoplasmic, and in the upper layers of the cortex—then LATE-NC should be ruled out. That is because so far, only Type A inclusions have been detected in cases of LATE-NC, whereas any of five morphological subtypes of TDP-43 pathology—types A to E—can be found in cases of FTLD. “If it’s not Type A, I would call it FTLD-TDP,” she said. When cases are difficult to call, Neumann noted that antibodies specific for different subtypes of TDP-43 pathology could come in handy. At the workshop, she showed preliminary data from two such antibodies—one that bound only Type A TDP-43, and one that bound all other types.
Clinical Relevance
Neumann also thinks that LATE-NC diagnoses need to be clinically relevant. As of now, a single TDP-43 inclusion in the amygdala would be grounds for diagnosing stage 1 LATE-NC. In a person with dementia, this scant level of pathology could not account for their decline. This problem harkens back to the early days of AD neuropathology research, Neumann said, when some might have considered a single amyloid plaque a sign of the disease. Neumann believes the staging criteria are too simplistic, and that the field needs more quantitative specificity that further defines both the neuropathological stages and their contributions to cognitive symptoms.
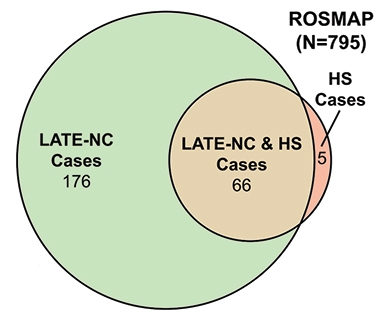
HS and LATE-NC. The vast majority of HS cases occurred in the context of LATE-NC, shown here in the ROS-MAP cohort. [Courtesy of Dugan et al., Neurology, 2021]
Nelson and others agreed that such updates to the LATE-NC criteria will be crucial moving forward, especially as more neuropathologists become familiar with this new category.
Overlap with TDP-43 pathology in other diseases has complicated the interpretation of LATE. For example, since the 2019 consensus report, scientists have delved into the relationship between LATE-NC and hippocampal sclerosis. Although the latter is commonly seen along with LATE-NC, it is not tied to any particular disease, and has been seen in people with other neurodegenerative diseases including AD and FTLD, as well as in the wake of neurological insults such as epilepsy and stroke. One study examining samples of more than 1,400 brains from the National Alzheimer’s Coordinating Center (NACC) and ROS-MAP cohorts, found that 66 of 77 cases of HS also had LATE-NC (Dugan et al., 2021; see image above). Another study examined samples from more than 400 brains in the NACC, finding that among 366 people with LATE-NC, 145 who also had hippocampal sclerosis trended toward having been more cognitively impaired during life, and tended to have more widespread TDP-43 pathology (Gauthreaux et al., 2022). The findings suggest that hippocampal sclerosis may represent a more advanced stage of LATE-NC, although the exact mechanisms linking the two remain unclear, Nelson said.
How Common is LATE?
Nelson presented fresh findings from the largest study on this question done to date. Because most research and clinic-based autopsy cohorts skew toward people with neurodegenerative disease, Nelson sought samples that were more representative of the general population. In all, he compiled autopsy data from 13 community- or population-based cohorts, comprising samples from 6,251 brains. Participants were an average of 88 years old when they died, and a quarter were ApoE4 carriers. Nelson reported that 39 percent of them had LATE-NC. Of those, a third were in LATE-NC stage 1, with TDP-43 pathology confined to the amygdala region. About a quarter of people across these cohorts had stage 2 or 3 LATE-NC, suggesting it could contribute to cognitive impairment.

LATE-NC Without AD. Among people who had no substantial AD pathology, more than a quarter had LATE-NC. [Courtesy of Peter Nelson, University of Kentucky.]
Nelson also examined relationships between AD pathology—in this case, the CERAD neuritic amyloid plaque score—and LATE-NC. Two-thirds of cases had CERAD scores indicative of at least some AD pathology, and about half of these also had LATE-NC. Of the one-third of participants whose low CERAD scores indicated little to no AD pathology, 27 percent had LATE-NC. The findings suggest that while LATE-NC is most prevalent among people with AD pathology, it also occurs on its own (see image below).
Nelson pointed out that, so far, studies suggest that LATE-NC on its own packs less of a wallop on cognition than does AD, resulting in a slower course of decline (Boyle et al., 2017; Nag et al., 2017). However, he noted that pure LATE-NC and pure AD are uncommon—far more often, both pathologies accumulate in a person's brain, resulting in a faster course of disease than either alone. Nelson said that biomarkers are desperately needed to diagnose LATE-NC, to study its natural history and role in cognitive decline, and to better plan and test outcomes in clinical trials (see Part 2 of this series).—Jessica Shugart
References
News Citations
- New Ubiquitinated Inclusion Body Protein Identified
- Introducing LATE—A Common TDP-43 Proteinopathy that Strikes After 80
- Across Time and Space: Machine Learning Reveals Paths to Dementia
- Forget Typical Alzheimer's: AI Finds Four Types.
Paper Citations
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007 Jul;171(1):227-40. PubMed.
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007 May;61(5):435-45. PubMed.
- James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain. 2016 Sep 30; PubMed.
- Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T, Dugger BN, Ghetti B, Grossman M, Hasegawa M, Herrup K, Holton J, Jellinger K, Lashley T, McAleese KE, Parisi JE, Revesz T, Saito Y, Vonsattel JP, Whitwell JL, Wisniewski T, Hu W. LATE to the PART-y. Brain. 2019 Sep 1;142(9):e47. PubMed.
- Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E, Van Deerlin VM, Abner EL, Jicha GA, Barber JM, Lee VM, Lee EB, Trojanowski JQ, Nelson PT. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain. 2020 Sep 1;143(9):2844-2857. PubMed.
- Dugan AJ, Nelson PT, Katsumata Y, Shade LM, Boehme KL, Teylan MA, Cykowski MD, Mukherjee S, Kauwe JS, Hohman TJ, Schneider JA, Alzheimer’s Disease Genetics Consortium, Fardo DW. Analysis of genes (TMEM106B, GRN, ABCC9, KCNMB2, and APOE) implicated in risk for LATE-NC and hippocampal sclerosis provides pathogenetic insights: a retrospective genetic association study. Acta Neuropathol Commun. 2021 Sep 15;9(1):152. PubMed.
- Gauthreaux KM, Teylan MA, Katsumata Y, Mock C, Culhane JE, Chen YC, Chan KC, Fardo DW, Dugan AJ, Cykowski MD, Jicha GA, Kukull WA, Nelson PT. Limbic-Predominant Age-Related TDP-43 Encephalopathy: Medical and Pathologic Factors Associated With Comorbid Hippocampal Sclerosis. Neurology. 2022 Apr 5;98(14):e1422-e1433. Epub 2022 Feb 4 PubMed.
- Boyle PA, Yang J, Yu L, Leurgans SE, Capuano AW, Schneider JA, Wilson RS, Bennett DA. Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain. 2017 Mar 1;140(3):804-812. PubMed.
- Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology. 2017 Feb 14;88(7):653-660. Epub 2017 Jan 13 PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.