Acetaminophen Derivative Tempers Microglia, Spurs Plaque Clearance
Quick Links
Researchers are searching for ways to dampen harmful inflammation and boost microglial phagocytosis in the Alzheimer’s brain. In the November 4 Proceedings of the National Academy of Sciences, researchers led by Hee Kyung Jin and Jae-sung Bae at Kyungpook National University, Daegu, and Mi Hee Lim at the Korea Advanced Institute of Science and Technology in Daejeon, both in the Republic of Korea, debut a small molecule, DAPPD, that seems to fit the bill. DAPPD resembles acetaminophen and enters the brain easily. In two mouse models of amyloidosis, chronic administration lightened amyloid load and preserved memory. Studies with cultured mouse and human cells found that DAPDD acts specifically on microglia, blocking the activation of the NLRP3 inflammasome. This lowered inflammatory signaling and stimulated phagocytosis.
- In mouse models, DAPPD lowered plaque load and improved memory.
- The compound switches microglia from an inflammatory to a phagocytic state.
- It inhibits the NLRP3 inflammasome.
Commenters found the study promising. “This manuscript is exciting since it shows a new small molecule that exerts therapeutic effects on two different AD murine models, both at the molecular and behavioral levels,” Giulio Pasinetti at the Icahn School of Medicine at Mount Sinai, New York, wrote to Alzforum (full comment below). Michael Heneka at the German Center for Neurodegenerative Diseases in Bonn, Germany, said the data jibe with other studies. “Together with previous observations, the present work by Park and colleagues further supports the hypothesis that NLRP3 inflammasome inhibition represents a valuable therapeutic target for the treatment of Alzheimer’s disease,” he wrote (full comment below).
Previous research had found that Aβ can activate the NLRP3 inflammasome, and that blocking this signaling lowers amyloidosis and bolsters memory in AD mice (Dec 2012 news). Several research groups are working on ways to target the NLRP3 inflammasome (Sep 2017 news).
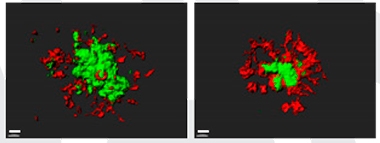
Feeding Frenzy. Microglia (red) from mice treated with DAPPD (right) cluster around amyloid plaques (green), devouring more Aβ than microglia in untreated mice (left). [Courtesy of Park et al., PNAS.]
The authors chose to test DAPPD, short for N,N'-diacetyl-p-phenylenediamine, based on its structural similarity to anti-inflammatory compounds. Like acetaminophen, DAPPD consists of a benzene ring, but it contains two acetamide groups instead of just one (see image below). First author Min Hee Park found that DAPPD had good drug properties. It was stable in plasma and did not bind plasma proteins. About 6 percent of DAPPD in the blood entered the brain.
Park and colleagues injected 2 mg/kg DAPPD intraperitoneally into 7.5-month-old APP/PS1 mice every day for a month. By 8.5 months, untreated controls had trouble remembering the location of a hidden platform in the Morris water maze, but treated mice performed like wild-type. After another month of treatment, the researchers sacrificed the mice and examined their brains. Treated mice had about one-sixth the amount of plaque in their hippocampi and cortices as did untreated controls. In a second mouse model, 5xFAD, which develops more aggressive amyloidosis, treatment of 3-month-old mice with DAPPD for one month cut plaque load in half and maintained memory at wild-type levels. The authors noted that the APP/PS1 study tested a prevention paradigm, because treatment started before plaques formed, while the 5xFAD experiment tested the ability of DAPPD to treat established disease.
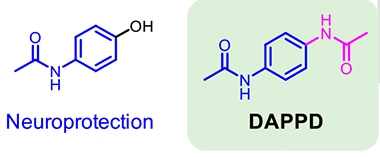
Tweaks to an Old Drug. DAPPD (right) differs from acetaminophen (left) by the addition of a second acetamide group (magenta). [Courtesy of Park et al., PNAS.]
The improvements correlated with dampened inflammation. In treated APP/PS1 mice, the amount of activated, Iba-positive microglia in cortex and hippocampus was about half that in untreated controls, while the amount of astrogliosis dropped by a third. Pro-inflammatory cytokines dropped to wild-type levels. In addition, more microglia clustered around plaques than in untreated mice (see image above). These microglia had a more rounded shape, characteristic of phagocytes, and harbored twice as much Aβ in their phagolysosomes as did microglia in untreated brains. Overall, the data suggested a switch from proinflammatory to a phagocytic phenotype.
To find out if DAPPD acted directly or indirectly on microglia, the authors treated cultures of mouse microglia, astrocytes, or neurons with synthetic Aβ42 to trigger inflammation. Adding DAPPD doused inflammation in the microglial cultures, but had no effect on the other two cell types, which continued to secrete signaling proteins that trigger microgliosis. In the presence of DAPPD, microglia expressed more homeostatic and anti-inflammatory genes, such as IL-4, TGF-β, and TMEM119, and less of several pro-inflammatory markers, including IL-6, TNFα, ApoE, and TREM2. Notably, conditioned media from microglia exposed to Aβ42 triggered inflammation in astrocytes, but not if the microglia had been treated with DAPPD.
How does DAPPD soothe microglia? In treated cells, the authors found it reduced protein components of the NLRP3 inflammasome, and suppressed cleavage of its downstream target caspase-1, a major pro-inflammatory protease. This inflammasome is controlled by the transcription factor NF-κB (Bauernfeind et al., 2009). Aβ boosts expression of NF-κB, and activates NLRP3. In mouse microglial cultures treated with Aβ, DAPPD treatment lowered NF-κB to wild-type levels. It had similar effects on a human microglial cell line, suppressing expression of NF-κB and NLRP3 inflammasome proteins and switching the cells from an inflammatory to a phagocytic state. Acetaminophen did not affect NF-κB in either murine or human microglial cells, confirming that DAPPD has a distinct mechanism of action.
In ongoing work, the authors are generating a chemical library to investigate the relationship between compound structure and activity, Lim wrote to Alzforum. They hope to find more potent compounds with even better medicinal properties than DAPPD.
Commenters were intrigued by the potential. “This group and others have advanced the field by focusing on a specific signaling pathway rather than just ‘microglial activation,’ and that is a huge step forward,” Malú Tansey at the University of Florida, Gainesville, wrote to Alzforum (full comment below). Still, she noted that many anti-inflammatory drugs have broad effects and can compromise the immune system. “Additional studies into inflammasome biology are warranted before these strategies can be deemed efficacious and safe,” Tansey wrote.
Pasinetti suggested confirming that DAPPD acts only through the inflammasome by testing it on microglia from NLRP3 inflammasome knockout mice, which are viable. He noted that the specific target of DAPPD remains to be identified. It is also unclear at what stage of disease the treatment would work best.
Greg Cole at the University of California, Los Angeles, said DAPPD’s mechanism of action is similar to that of curcumin, which he investigates (Nov 2001 news; Dec 2004 news). Curcumin also quenches pro-inflammatory cytokines while boosting anti-inflammatory ones. However, low doses of curcumin raise TREM2, rather than lowering it. Cole wondered whether DAPPD might also raise TREM2 under some conditions, for example in plaque-associated microglia (see comment below). Cole suggested investigating DAPPD, curcumin, and similar compounds in ApoE and tauopathy mouse models as well.—Madolyn Bowman Rogers
References
News Citations
- Microglia and AD—Does the Inflammasome Drive Aβ Pathology?
- New AD Target: Silencing the NLRP3 Inflammasome with Boron?
- Is It the Curry?
- Curry Ingredient Spices Things Up by Blocking Aβ Aggregation
Research Models Citations
Paper Citations
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009 Jul 15;183(2):787-91. Epub 2009 Jul 1 PubMed.
External Citations
Further Reading
Primary Papers
- Park MH, Lee M, Nam G, Kim M, Kang J, Choi BJ, Jeong MS, Park KH, Han WH, Tak E, Kim MS, Lee J, Lin Y, Lee YH, Song IS, Choi MK, Lee JY, Jin HK, Bae JS, Lim MH. N,N'-Diacetyl-p-phenylenediamine restores microglial phagocytosis and improves cognitive defects in Alzheimer's disease transgenic mice. Proc Natl Acad Sci U S A. 2019 Nov 19;116(47):23426-23436. Epub 2019 Nov 4 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Klinik und Poliklinik für Neurologie
This paper by Park and colleagues reports novel findings showing that N,N'-Diacetyl-p-phenylenediamine (DAPPD) reduces Aβ plaque burden along with micro- and astroglial reactivity in APP/PS1 transgenic mice. DAPPD treatment decreased inflammatory cytokine expression and promoted microglial Aβ clearance as evidenced through confocal imaging of LAMP1-positive, Aβ containing Iba-1-positive microglia, together resulting in improved learning and memory.
This phenomenon was associated with a negative regulation of NLRP3 inflammasome, which has previously been shown to modulate microglial Aβ phagocytosis in vivo (Heneka et al. 2013). In the present paper, DAPPD treatment of APP/PS1 animals decreased the expression of NLRP3 and ASC, two of members of NLRP3 inflammasome pathway, presumably through an inhibition of NF-κB signalling. Activation of the NLRP3 inflammasome requires a transcriptional NF-κB-dependent upregulation of NLRP3 and pro-forms of interleukin-1β and interleukin-18, which then lead to the formation of the NLRP3 inflammasome and subsequently to caspase-1-mediated cleavage into active cytokines (Heneka et al. 2018). Together with previous observations (Demsey et al., 2017; Feng et al., 2017), the present work by Park and colleagues further supports the hypothesis that NLRP3 inflammasome inhibition represents a valuable therapeutic target for the treatment of Alzheimer’s disease.
References:
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674-8. Epub 2012 Dec 19 PubMed.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018 Oct;19(10):610-621. PubMed.
Feng J, Wang JX, Du YH, Liu Y, Zhang W, Chen JF, Liu YJ, Zheng M, Wang KJ, He GQ. Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci Ther. 2018 Dec;24(12):1207-1218. Epub 2018 Jun 4 PubMed.
Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AA, Cooper MA, O'Neill LA, Lynch MA. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. 2017 Mar;61:306-316. Epub 2016 Dec 18 PubMed.
The University of Florida College of Medicine
Park and colleagues report that targeting the inflammasome with a small molecule mitigates inflammation and cognitive behavioral deficits in mouse models. This and other groups have advanced the field by focusing on a specific signaling pathway rather than just “microglia activation” and that is a huge step forward for the field.
While it’s clear that various strategies can shut down NLRP3 signaling, we do not know enough about inflammasome biology to confidently say that this approach will afford beneficial effects without compromising immune competency and/or induce immunosuppression in the host. There is a strong epidemiological history of “dirty” anti-inflammatory drugs, including ibuprofen and non-selective compounds, and several of these strategies unequivocally suppress the immune system and render hosts vulnerable to opportunistic infections. Therefore, additional studies are warranted into inflammasome biology before these strategies can be deemed efficacious and safe.
UCLA/VA
This very nice and extensively documented report demonstrates the efficacy of a new small molecule (DAPPD) that inhibits NF-κB pathway-mediated activation of microglia by Aβ to restore microglial amyloid clearance in two lines of APP/PS1 mutant mice. They have good compound pharmacokinetics and stability and show efficacy in vitro in human cell lines. DAPPD’s anti-inflammatory function depends on its acetamide groups, which is not surprising as many anti-inflammatory compounds use reactive carbonyls to target reactive thiols in the pro-inflammatory IκB Kinase upstream signaling to NF-κB. For example, these papers describe larger acetamide based IKKβ inhibitors (Gillooly et al., 2009; Haddad et al., 2001). The authors point out that DAPPD is attractively simple, small, and stable—and has good drug properties. This is an important study that documents the preclinical efficacy of a new, small-molecule, immunomodulatory alternative to anti-Aβ antibody approaches to promoting amyloid clearance by microglia, while limiting their neurodegenerative phenotype.
This approach is reminiscent of our work over the last two decades with curcumin, a very well-studied anti-neuroinflammatory compound that uses a beta di-ketone bridge to target NF-κB signaling (see Jobin et al., 1999, and subsequent reports). Thus, one suspects that DAPPD and curcumin share at least this common target and the property of improving microglial phagocytosis and amyloid clearance and it is interesting to make some comparisons. In our most recent paper on curcumin and microglial phagocytosis (Teter et al., 2019), we addressed this issue and noted dose-dependent efficacy of curcumin in AD model mice and in cultured human cells. We reported that low doses of curcumin reduced IL-1β, iNOS, TNFα, and increased M2 markers like Arg1. DAPPD has similar activity, but at the single dose used, it slightly lowers Trem2 in microglia isolated from cortex. In contrast, we found that low doses but not high doses of curcumin increased Trem2 expression in cortex. It remains unclear whether and when in the course of AD pathogenesis it is helpful to have more or less Trem2—and in what subset of microglia.
When you do in vivo experiments and look some months after treatment, you may not see the initial activation and clearance but instead see the results of having reduced the numbers of plaques and Aβ—the causes of microglial activation. Thus, while there may be differences in microglia phenotype modulation between DAPPD and curcumin, these are also likely dose-dependent. Another possible explanation for the difference in TREM2 is that our data focused on increases in Trem2 on phosphotyrosine positive-phagocytic microglia associated with plaques while this paper didn’t seem to discriminate plaque-associated from bulk microglia. Since the treatments reduce the number of plaques, they likely also reduce plaque-associated microglia, which may result in lower levels of expression of mRNA elevated in plaque-associated microglia in the bulk isolated microglia pool. That is, as De Strooper’s recent microglial transcriptomics paper shows, there are a number of different types of microglia and only a small number of the bulk microglia have the high expression of GPNMB/Clec7a or Trem2 in the disease-associated (DAM) or neurodegenerative phenotypes associated with plaque microglia. We normalized expression of TREM2 with another AD gene microglial marker, CD33, whose higher level expression has been genetically implicated in increasing AD risk and lower levels in reducing AD risk, and we found that curcumin increased the TREM2/CD33 ratio. It may be that DAPPD does the same thing.
Whether or not it is desirable to increase or decrease TREM2, we also reported at AAIC that the more bioavailable curcumin formulation we have in clinical trials can reduce TREM2, Clec7a, and GPMNB in vivo while reducing pro-inflammatory cytokines and GFAP. The big disadvantage of curcumin is that it is less stable, but when adequately formulated we can get human plasma levels equivalent to the effective levels we and others find in animal models, and the limited bioavailability makes it difficult to overdose. The big advantages of curcumin are that it has a long history of safe use and is much less expensive than a new drug is likely to be. It will be very interesting to see how well these compounds perform in other animals with ApoE4 and tauopathy.
References:
Gillooly KM, Pattoli MA, Taylor TL, Chen L, Cheng L, Gregor KR, Whitney GS, Susulic V, Watterson SH, Kempson J, Pitts WJ, Booth-Lute H, Yang G, Davies P, Kukral DW, Strnad J, McIntyre KW, Darienzo CJ, Salter-Cid L, Yang Z, Wang-Iverson DB, Burke JR. Periodic, partial inhibition of IkappaB Kinase beta-mediated signaling yields therapeutic benefit in preclinical models of rheumatoid arthritis. J Pharmacol Exp Ther. 2009 Nov;331(2):349-60. Epub 2009 Aug 3 PubMed.
Haddad JJ, Safieh-Garabedian B, Saadé NE, Land SC. The biphasic immunoregulation of pyrimidylpiperazine (Y-40138) is IL-10 sensitive and requires NF-kappa B targeting in the alveolar epithelium. Br J Pharmacol. 2001 May;133(1):49-60. PubMed.
Jobin C, Bradham CA, Russo MP, Juma B, Narula AS, Brenner DA, Sartor RB. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J Immunol. 1999 Sep 15;163(6):3474-83. PubMed.
Teter B, Morihara T, Lim GP, Chu T, Jones MR, Zuo X, Paul RM, Frautschy SA, Cole GM. Curcumin restores innate immune Alzheimer's disease risk gene expression to ameliorate Alzheimer pathogenesis. Neurobiol Dis. 2019 Jul;127:432-448. Epub 2019 Apr 2 PubMed.
Icahn School of Medicine at Mount Sinai
Icahn School of Medicine at Mount Sinai
Park et al. describe a new small molecule targeting the NF-κβ pathway that is able to restore microglia dysfunction, improve cognitive impairment, and foster Aβ clearance in two mouse models of Alzheimer’s disease. The authors showed that N,N'-Diacetyl-p-phenylenediamine (DAPPD) is metabolically stable, reaches the brain at a physiological concentration, and has optimal pharmacokinetic properties. Regarding the properties of DAPPD, the authors demonstrate in an extensive study that includes in vivo research using two different mouse models of AD that the small compound improves spatial learning and memory, decreases Aβ aggregate deposition, and restores glial activation through the reduction of pro-inflammatory cytokines and the promotion of anti-inflammatory ones. They confirmed these results using human microglia cultures, and additionally they showed that DAPPD restores the phagocytic capacity that Aβ-treated microglia had lost. Exploring deeper into the mechanism of action, they hypothesized that the therapeutic effects of this small molecule are due to the downregulation of proteins conforming the NLRP3 inflammasome —NLRP3, ASC and IL-1β— through the suppression of the NF-κβ pathway. Thus, they propose that by impacting this pathway, DAPPD reduces NLRP3-mediated neuroinflammation, allowing microglia to recover their phenotype and phagocytic capacity, which finally allows microglia to clear larger Aβ aggregates, improving the cognitive capabilities of AD mice.
This manuscript is exciting since it shows a new small molecule that exerts therapeutic effects on two different AD murine models, both at the molecular and behavioral levels. It proposes a double approach to ameliorate the disease, consisting on the one hand of decreasing amyloidosis by restoring the phagocytic aptitude of microglia; and on the other hand by decreasing neuroinflammation through inhibition of the NF-κβ pathway, which re-establishes the homeostatic balance between pro- and anti-inflammatory cytokines.
DAPPD appears as a promising therapeutic agent, however, some more questions should be addressed to provide a more complete picture of the mechanism of action of this molecule. For instance, the authors claim the DAPPD beneficial effects are due to reduced expression of proteins conforming the NLRP3 inflammasome, although they do not validate that hypothesis by demonstrating, for instance, reduced effect of the drug on NLRP3 knockout microglia. Moreover, they have evidence that DAPPD impacts the NF-κβ pathway, but the specific target in this pathway remains to be identified. Finally, it also would need to be clarified whether the restoration of the microglial phagocytic ability is a consequence of this same pathway, or of other mechanisms and a different target that is involved in this process. Collectively, the paper might help to clarify the potential role of inflammation through inflammasome-mediated responses, whose regulation by DAPPD may influence amyloid pathology. Thus, it is possible that a coordinated response in inflammatory homeostasis may attenuate β-amyloidosis. Still, the question of whether preclinical or prodromal stage of disease should be targeted with this nevertheless exciting compound remains unanswered.
Boston University Chobanian & Avedisian School of Medicine
A few months ago, some people were ready to close the book on NSAIDs based on the results of the INTREPAD trial led by John Breitner at McGill, which found that daily naproxen use by asymptomatic people in their 60s at high risk for Alzheimer’s disease did not improve cognition or CSF markers, and may have even made the primary outcome a bit worse.
At that time I and others opined that those results did not debunk any of the potential benefits mediated by reducing neuroinflammation, especially if this could be achieved over the course of a lifetime. Classic NSAIDs and other anti-inflammatory agents differ in their ability to suppress NF-kappaB activation (Takada et al., 2004). By building a better mouse trap, these researchers have demonstrated that the inflammasome remains a viable target.
This may prove to be especially true if this pathway is targeted earlier, with compounds that carry low risk for toxicity with long-term use such as resveratrol which has already been shown to be safe and well-tolerated (Turner et al., 2015). The naturally occurring neurosteroid allopregnanolone also tamps down NFkB activation. More work may be needed, but the inflammasome book should definitely not be closed.
References:
Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene. 2004 Dec 9;23(57):9247-58. PubMed.
Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA, Brewer JB, Rissman RA, Raman R, Aisen PS, Alzheimer's Disease Cooperative Study. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology. 2015 Oct 20;85(16):1383-91. Epub 2015 Sep 11 PubMed.
Make a Comment
To make a comment you must login or register.