Commander Complexes in Lysosomes Stave Off Parkinson’s Risk
Quick Links
Evidence suggests that in Parkinson’s disease, a breakdown in lysosomal function allows lipids to build up within cells. Variants of the GBA1 gene, the strongest genetic risk factor for sporadic PD, are partly to blame. GBA1 encodes the glucocerebrosidase enzyme that helps process lipids. But GBA1 variants alone do not fully explain reduced GCase activity in PD. Now, scientists led by Dimitri Krainc of Northwestern University Feinberg School of Medicine, Chicago, report that another gene family might be involved. In the April 11 Science, they describe a link between GCase activity and the Commander complex, which helps recycle endosomal cargo to the plasma membrane.
- GBA1 mutations alone do not explain lysosomal dysfunction in PD.
- Loss of endosomal recycling proteins cripple this lysosomal enzyme.
- Without a protein called COMMD3, cells spit out would-be lysosomal proteins in exosomes.
“Our data found a new function of this complex,” Krainc told Alzforum. Previously, he and collaborators had described how GCase dysfunction leads to α-synuclein accumulation, and vice versa, in PD neurons. This helped explain the connection between PD and Gaucher’s disease, a childhood lysosomal lipid storage disorder also caused by GBA1 mutation (see Mazzulli et al., 2011).
The pathways that determine these different disease outcomes are still ill-defined, noted David Sulzer of Columbia University Medical Center, New York, who was not involved in either study. “This is a nice study that has identified a means by which lysosomal cargo may be differentially handled. The novel approaches enabled the identification of many different proteins that modulate lysosomal organelle function and allowed the investigators to focus on a particular pathway that regulates many trafficking steps and components,” he added.
To identify genes other than GBA1 that affect lysosomal GCase activity, first author Georgia Minakaki and colleagues ran a genome-wide CRISPR interference screen. This revealed 338 genes that may impact lysosomal GCase function. Of the 190 genes that decreased activity, the top hit goes by the mouthful “copper metabolism MURR1 domain-containing 3.” COMMD3 and the other members of the COMMD family combine with trafficking proteins called CCDC22 and CCDC93 to form a 12-protein complex dubbed CCC. This is one component of the Commander complex. Others are Retriever, WASH, and DENND10, all of which participated in endosomal recycling independently of the retromer complex.
The authors found that knocking out COMMD3 halved lysosomal GCase activity in human embryonic kidney cells and dampened lysosomal GCase in microglia and neurons derived from human iPSCs. It also compromised CCC complexes, leaving them short of CCDC22. Adding back COMMD3 to these cells rescued COMMD1 formation and CCDC2 expression as well as GCase activity.
Isolating endolysosomes revealed significant differences in lysosomal protein levels between COMMD3 knockout and wild-type cells. The researchers measured nearly 40 percent less LAMP2, LIMP-2, GCase, and cathepsin B in the knockout cell lysosomes. These cells also cleaved prosaposin and progranulin less effectively. Progranulin processing helps preserve lysosomal function, while prosaposin helps these organelles metabolize lipids (see Simon et al., 2022; Meyer et al., 2014).
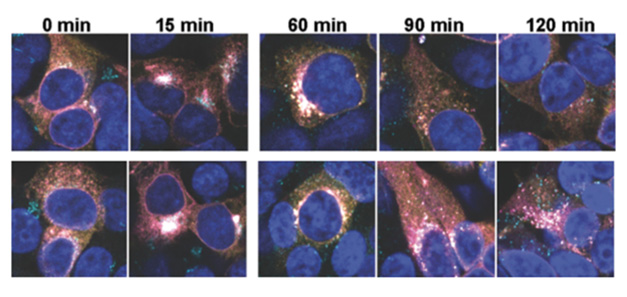
Endosome Encapsulated. After leaving the Golgi (turquoise dots in the 0- and 15-minute columns), GCase (yellow) and LIMP2 (magenta) co-localize with endosomes (turquoise in the 60- to 120-minute columns) in COMM3 knockout (bottom row) but not in wild-type HEK cells (top row). [Courtesy of Minakaki et al., Science, 2025.]
Since the CCC and Commander complexes are both involved in endosomal recycling, the researchers wondered whether they help ferry lysosomal proteins to their destination. They tracked LIMP-2 and GCase in HEK cells. In COMMD3 knockouts, these proteins were captured by post-Golgi vesicles, some of which appeared to be early endosomes (image above). Examining extracellular vesicles from wild-type and knockout cells, Minakaki found more of the lysosomal proteins LAMP1, LIMP-2, and HEXB in the latter, suggesting that, at least in this cell line, these proteins are trafficked out of the cell rather than into lysosomes when there is no COMMD3. Similarly, extracellular vesicles from iPSC-derived neurons lacking COMMD3 contained 12 times as much LAMP1 and three times as much HEXB as did extracellular vesicles from wild-type neurons. All told, the authors interpret the findings to suggest that the CCC and Commander complexes influence the fate of lysosomal proteins.
To investigate further, the authors searched genomics data for links between PD and protein variants in these complexes. In the U.K. Biobank whole-exome cohort and in the whole-genome AMP-PD cohort—a total of 6,166 PD cases and 109,467 controls—they found that loss-of-function variants in the COMMD family, the CCC complex, or the Commander complex combined were at least twice as common in PD as in controls. The one individual gene with a significant effect was COMMD9, but only in the U.K. Biobank cohort.
The authors acknowledge that since they are required to traffic hundreds of plasma membrane proteins, loss-of-function variants in these complexes may be extremely rare. A larger sample size might offer a better chance of spotting links between individual commander complex proteins and PD, DLB, and other neurodegenerative diseases, they wrote.—Lauren Schneider
Lauren Schneider is a freelance writer in New York City.
References
Paper Citations
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011 Jul 8;146(1):37-52. Epub 2011 Jun 23 PubMed.
- Simon MJ, Logan T, DeVos SL, Di Paolo G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. 2022 Oct 13; PubMed.
- Meyer RC, Giddens MM, Coleman BM, Hall RA. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014 Oct 17;1585:1-12. Epub 2014 Aug 15 PubMed.
Further Reading
Primary Papers
- Minakaki G, Safren N, Bustos BI, Lubbe SJ, Mencacci NE, Krainc D. Commander complex regulates lysosomal function and is implicated in Parkinson's disease risk. Science. 2025 Apr 11;388(6743):204-211. Epub 2025 Apr 10 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Columbia University
This nice study identified a means by which lysosomal cargo may be differentially handled. The novel approaches enabled the identification of many different proteins that modulate lysosomal organelle function, and allowed the investigators to focus on a particular pathway that regulates many trafficking steps and components in lysosomes. The observation that secretory organelles related to lysosomes can undergo membrane fusion has been conveyed since the original studies by De Duve and associates (Novikoff et al., 1956). It has become more central to the study of neurodegenerative disorders, in part due to the identification of a nominally lysosomal cargo enzyme in familial PD. This will continue to be an exciting direction for study of intracellular communication and stress response pathways.
The PD field lacks clarity on whether pathogenesis related to many, but not all mutations of GBA—one of multiple lysosomal cargos likely to be modified by this pathway—is related to a decrease in activity within the lysosomal lumen, as modeled in this study, or rather to issues with protein mishandling that may occur on the cytosolic face of the lysosome, as originally reported for GBA mutants in Gaucher’s disease by Mia Horowitz (see Horowitz et al., 2022). Neither theory, nor the present study, is entirely satisfactory in explaining the differences between Gaucher’s and Parkinson’s caused by mutations in the same gene and its relationship with α-synuclein accumulation.
These relationships will be important for understanding, among other things, why the pathways lead to death of particular neuronal populations in PD and other symptoms in Gaucher’s. The cellular steps involved are obviously important if the disorder is to be treated by overexpression of GBA, as has been successful for Gaucher’s, or by targeting regulatory pathways of lysosomal trafficking, such as the complex recommended in this study, or LIMP1, or by pathways that regulate other steps, such as response to misfolded proteins.
Regardless of the specific step by which GBA mutants lead to PD, regulating lysosomal content and membrane fusion are central to cell biology and will be important for many addressing many questions about normal function and disease. This study has made a genuine contribution to these research directions.
References:
NOVIKOFF AB, BEAUFAY H, DE DUVE C. Electron microscopy of lysosomerich fractions from rat liver. J Biophys Biochem Cytol. 1956 Jul 25;2(4 Suppl):179-84. PubMed.
Horowitz M, Braunstein H, Zimran A, Revel-Vilk S, Goker-Alpan O. Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease. Adv Drug Deliv Rev. 2022 Aug;187:114402. Epub 2022 Jun 25 PubMed.
Make a Comment
To make a comment you must login or register.