Defining Biomarker Curves, Progression Models for Familial FTD
Quick Links
In the Alzheimer’s field, detailed knowledge of how biomarkers change at preclinical stages has enabled researchers to select the best participants for disease-modifying prevention trials. For frontotemporal dementia, this knowledge has been lacking—until now. In the September 22 Nature Medicine, a large international collaboration led by Adam Staffaroni and Adam Boxer at the University of California, San Francisco, detail how two markers of neurodegeneration, brain atrophy and plasma neurofilament light (NfL), increase in FTD mutation carriers starting as many as 40 years before they have symptoms. The onset, speed, and even order of biomarker change varied according to whether a person had a C9ORF72, GRN, or MAPT genetic variant. In a given mutation carrier, scientists could predict the age when symptoms would appear within about five years. Knowing estimated onset ages would greatly reduce the number of people needed for prevention studies, the authors calculated. This will make trials of these rare conditions more feasible, Staffaroni said.
- Data from familial FTD cohorts reveal patterns of biomarker and clinical change.
- C9ORF72, GRN, and MAPT mutations evoke distinct biomarker changes.
- Progression models could help select participants for prevention trials.
Other scientists welcomed the data. “This is a fantastic and long overdue study,” Michael Gold at AbbVie wrote to Alzforum. “For the first time, we have data on reasonably large groups of patients with genetically defined forms of FTD, and can directly track changes on biomarkers and clinical endpoints over time,” he wrote (comment below).
Dan Geschwind at the University of California, Los Angeles, agreed that the paper advances the field. “It is a beautiful example of how coordinated, highly collaborative research efforts are necessary to move the needle,” he said (comment below). A co-author, Geschwind runs the genetics core for one of the FTD cohorts analyzed in the paper.
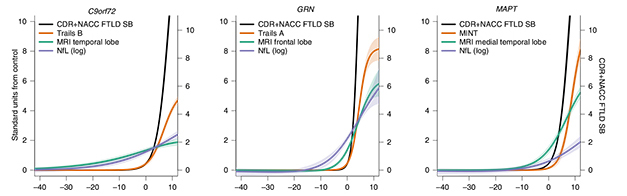
Progression Curves. Cross-sectional data predicts that in C9ORF72 carriers (left), temporal lobe atrophy (green) and serum NfL (purple) diverge from controls 30 years or more before symptom onset (x axis), but change slowly. In GRN carriers (middle), NfL climbs 10 years before frontal lobe atrophy does, but both rise swiftly. In MAPT carriers (right), medial temporal lobe atrophy and NfL rise about 10 years prior to symptom onset, with atrophy having a much steeper trajectory. In all carriers, clinical status (black) and specific cognitive test scores (orange) worsen rapidly after onset. Which cognitive domain is most affected also varies by mutation. [Courtesy of Staffaroni et al., Nature Medicine.]
Tackling a Heterogeneous Disease
Frontotemporal dementia studies are complicated because it is a syndrome that can have several underlying causes with distinct pathologies. Almost a third of cases have a strong genetic basis and run in families, with most due to mutations in the MAPT gene for tau, in the GRN gene for progranulin, or in C9ORF72 (Nov 2014 conference news; Greaves and Rohrer, 2019). However, even within affected families the disease is unpredictable, with symptoms appearing at different ages (Moore et al., 2020). This heterogeneity has made it challenging to design effective prevention trials.
To tackle this, scientists launched the FTD Prevention Initiative, an umbrella group that brings together several large familial FTD cohort studies from around the world (Mar 2021 conference news; Mar 2021 conference news). For the current study, the authors combined data from the two largest cohorts, ALLFTD in North America and GENFI in Europe and Canada. They compared how biomarkers and clinical scores changed as disease progressed in people with a mutation in any of these three genes. Out of 796 carriers, 347 had a C9ORF72 expansion, 281 had a GRN mutation, and 168 had a MAPT mutation. The study also included 412 noncarrier family members as controls.
Most participants had undergone volumetric MRI and donated blood; all took cognitive and neuropsychological tests. The key clinical measure used was the Clinical Dementia Rating plus National Alzheimer’s Coordinating Center Frontotemporal Lobar Degeneration Module (CDR+NACC-FTLD). This test combines the six domains of the standard CDR with two additional domains focused on behavior and language, which tend to be affected in FTD. As with the standard CDR, a global score of 0 indicates normal cognition, and 3 maximum impairment. A sum-of-boxes score, similar to that used in AD, ranges from 0 to 24 (Miyagawa et al., 2020; Mar 2021 conference news).
Three Genes, Three Distinct Profiles
In cross-sectional data, the findings were strikingly different for each mutation. In both cohorts, C9ORF72 carriers had the earliest biomarker changes. Already in early adulthood, as much as 40 years before symptom onset, multiple brain regions were smaller compared with those same regions in controls. Previous studies have seen this as well, and suggested it could represent a developmental deficit (Lee et al., 2017; Caverzasi et al., 2019). Atrophy worsened over time, with the volume of temporal and parietal brain regions dropping one standard deviation below that of controls about six years before symptom onset. Likewise, plasma NfL rose early, diverging from controls as much as 30 years prior to disease onset, and reaching one standard deviation three years prior.
GRN mutation carriers were affected somewhat later. Their frontal and temporal regions began shrinking up to 10 years prior to symptom onset, reaching one standard deviation one year before. However, in contrast to the other carriers, NfL levels rose before atrophy did. NfL diverged from controls 15 years before symptom onset, and reached one standard deviation five years prior.
Effects showed up latest in MAPT carriers. Most pronounced in the medial temporal lobe, brain atrophy showed up 10 years before symptom onset, reaching one standard deviation higher two years prior. Other brain regions only began to decline after symptoms appeared. NfL did not rise until symptom onset, and only reached one standard deviation higher five years later.
Longitudinal data added information on the rates of change. Here, GRN carriers stood out as declining most rapidly. After symptom onset, they lost brain volume four times faster than C9ORF72 carriers, and about twice as fast as MAPT carriers. Their NfL rose three times as quickly as in the other carriers. Likewise, their clinical status declined more steeply after symptoms began, losing about two points on the CDR+NACC-FTLD-SB per year, compared to 1.45 points for MAPT carriers and 1.25 for C9ORF72 carriers.
The findings largely match those of previous studies, which showed mutation-specific differences and flagged GRN mutations as causing the most aggressive disease (Whitwell et al., 2015; Mar 2021 conference news). However, Staffaroni noted this is the first comprehensive effort that combines cross-sectional and longitudinal clinical, imaging, and plasma biomarker data across consortia to systematically compare the three major genetic causes of FTD head-to-head. Importantly, the findings for each mutation were consistent across the two cohorts on different continents, which Charlotte Teunissen at Amsterdam University Medical Center called remarkable (see comment below). This will make it easier to run international prevention trials, since selection criteria and outcome measures can be uniform across the studies.
Designing More Efficient Trials
How can this information be used to enable those trials? The authors integrated biomarker and clinical data from the progression curves to calculate each participant’s “disease age,” i.e., estimated age of onset, and assessed the accuracy of these predictions. As might be expected, biomarker and clinical data taken from very early in the disease course gave estimates that were off by about 15 years. Within 10 years of symptoms, however, the progression models were able to estimate disease age within 5.5 years, close to the accuracy in familial AD (Oxtoby et al., 2018).
The authors calculated that a hypothetical four-year prevention trial using MRI as an outcome measure would need to enroll 394 C9ORF72 carriers, 459 GRN carriers, or 526 MAPT carriers to detect a 50 percent slowing of disease. However, if they selected participants within 2.5 years of their disease age, these numbers came down to 102, 24, and 24, respectively. The number is higher for C9ORF72 carriers because of their relatively slower decline on MRI. For GRN and MAPT, the numbers are low enough that participants could be recruited from existing longitudinal cohorts. “Prevention trials are feasible, but will likely require a global recruitment effort,” Staffaroni said.
In addition to selecting participants, progression models could be used to choose the best outcome measure for each mutation, Staffaroni said. Most current FTD trials test a mutation-specific intervention, such as an anti-sortilin antibody to boost progranulin levels in GRN carriers, or antisense oligonucleotides that target the expanded C9ORF72 transcript (Aug 2021 conference news; Mar 2021 news). The models could also help predict progression rates and variability, or even be used to simulate changes in untreated mutation carriers to enrich placebo groups. Smaller control groups would aid recruitment for trials, since people are likeliest to sign up if they know they have a higher chance to be on drug. Staffaroni said the next step is to talk with regulatory agencies about how to use these models to design registration trials.—Madolyn Bowman Rogers
References
News Citations
- Stream of Genetics Pushes FTD Research Forward
- Cohorts Band Together to Get Global FTD Trials Going
- Merged Consortia Forge Path to Trials in Frontotemporal Dementia
- Moving Target: Can Standardized Tests Track Symptoms of FTD?
- Imaging Exposes Hugely Heterogeneous Brain Changes Among FTDs
- AL001 Boosts Progranulin. Does it Slow Frontotemporal Dementia?
- FTD Trials: The Now and the Future
Paper Citations
- Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019 Aug;266(8):2075-2086. Epub 2019 May 22 PubMed.
- Moore KM, Nicholas J, Grossman M, McMillan CT, Irwin DJ, Massimo L, Van Deerlin VM, Warren JD, Fox NC, Rossor MN, Mead S, Bocchetta M, Boeve BF, Knopman DS, Graff-Radford NR, Forsberg LK, Rademakers R, Wszolek ZK, van Swieten JC, Jiskoot LC, Meeter LH, Dopper EG, Papma JM, Snowden JS, Saxon J, Jones M, Pickering-Brown S, Le Ber I, Camuzat A, Brice A, Caroppo P, Ghidoni R, Pievani M, Benussi L, Binetti G, Dickerson BC, Lucente D, Krivensky S, Graff C, Öijerstedt L, Fallström M, Thonberg H, Ghoshal N, Morris JC, Borroni B, Benussi A, Padovani A, Galimberti D, Scarpini E, Fumagalli GG, Mackenzie IR, Hsiung GR, Sengdy P, Boxer AL, Rosen H, Taylor JB, Synofzik M, Wilke C, Sulzer P, Hodges JR, Halliday G, Kwok J, Sanchez-Valle R, Lladó A, Borrego-Ecija S, Santana I, Almeida MR, Tábuas-Pereira M, Moreno F, Barandiaran M, Indakoetxea B, Levin J, Danek A, Rowe JB, Cope TE, Otto M, Anderl-Straub S, de Mendonça A, Maruta C, Masellis M, Black SE, Couratier P, Lautrette G, Huey ED, Sorbi S, Nacmias B, Laforce R Jr, Tremblay ML, Vandenberghe R, Damme PV, Rogalski EJ, Weintraub S, Gerhard A, Onyike CU, Ducharme S, Papageorgiou SG, Ng AS, Brodtmann A, Finger E, Guerreiro R, Bras J, Rohrer JD, FTD Prevention Initiative. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020 Feb;19(2):145-156. Epub 2019 Dec 3 PubMed.
- Miyagawa T, Brushaber D, Syrjanen J, Kremers W, Fields J, Forsberg LK, Heuer HW, Knopman D, Kornak J, Boxer A, Rosen HJ, Boeve BF, Appleby B, Bordelon Y, Bove J, Brannelly P, Caso C, Coppola G, Dever R, Dheel C, Dickerson B, Dickinson S, Dominguez S, Domoto-Reilly K, Faber K, Ferrell J, Fishman A, Fong J, Foroud T, Gavrilova R, Gearhart D, Ghazanfari B, Ghoshal N, Goldman JS, Graff-Radford J, Graff-Radford N, Grant I, Grossman M, Haley D, Hsiung R, Huey E, Irwin D, Jones D, Jones L, Kantarci K, Karydas A, Kaufer D, Kerwin D, Kraft R, Kramer J, Kukull W, Litvan I, Lucente D, Lungu C, Mackenzie I, Maldonado M, Manoochehri M, McGinnis S, McKinley E, Mendez MF, Miller B, Multani N, Onyike C, Padmanabhan J, Pantelyat A, Pearlman R, Petrucelli L, Potter M, Rademakers R, Ramos EM, Rankin K, Rascovsky K, Roberson ED, Rogalski E, Sengdy P, Shaw L, Tartaglia MC, Tatton N, Taylor J, Toga A, Trojanowski JQ, Wang P, Weintraub S, Wong B, Wszolek Z. Utility of the global CDR® plus NACC FTLD rating and development of scoring rules: Data from the ARTFL/LEFFTDS Consortium. Alzheimers Dement. 2020 Jan;16(1):106-117. PubMed.
- Lee SE, Sias AC, Mandelli ML, Brown JA, Brown AB, Khazenzon AM, Vidovszky AA, Zanto TP, Karydas AM, Pribadi M, Dokuru D, Coppola G, Geschwind DH, Rademakers R, Gorno-Tempini ML, Rosen HJ, Miller BL, Seeley WW. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. 2017;14:286-297. Epub 2016 Dec 10 PubMed.
- Caverzasi E, Battistella G, Chu SA, Rosen H, Zanto TP, Karydas A, Shwe W, Coppola G, Geschwind DH, Rademakers R, Miller BL, Gorno-Tempini ML, Lee SE. Gyrification abnormalities in presymptomatic c9orf72 expansion carriers. J Neurol Neurosurg Psychiatry. 2019 Sep;90(9):1005-1010. Epub 2019 May 11 PubMed.
- Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC, DeJesus-Hernandez M, Knopman DS, Wszolek ZK, Petersen RC, Rademakers R, Jack CR Jr, Josephs KA. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. 2015 May;22(5):745-52. Epub 2015 Feb 12 PubMed.
- Oxtoby NP, Young AL, Cash DM, Benzinger TL, Fagan AM, Morris JC, Bateman RJ, Fox NC, Schott JM, Alexander DC. Data-driven models of dominantly-inherited Alzheimer's disease progression. Brain. 2018 May 1;141(5):1529-1544. PubMed.
Further Reading
Primary Papers
- Staffaroni AM, Quintana M, Wendelberger B, Heuer HW, Russell LL, Cobigo Y, Wolf A, Goh SM, Petrucelli L, Gendron TF, Heller C, Clark AL, Taylor JC, Wise A, Ong E, Forsberg L, Brushaber D, Rojas JC, VandeVrede L, Ljubenkov P, Kramer J, Casaletto KB, Appleby B, Bordelon Y, Botha H, Dickerson BC, Domoto-Reilly K, Fields JA, Foroud T, Gavrilova R, Geschwind D, Ghoshal N, Goldman J, Graff-Radford J, Graff-Radford N, Grossman M, Hall MG, Hsiung GY, Huey ED, Irwin D, Jones DT, Kantarci K, Kaufer D, Knopman D, Kremers W, Lago AL, Lapid MI, Litvan I, Lucente D, Mackenzie IR, Mendez MF, Mester C, Miller BL, Onyike CU, Rademakers R, Ramanan VK, Ramos EM, Rao M, Rascovsky K, Rankin KP, Roberson ED, Savica R, Tartaglia MC, Weintraub S, Wong B, Cash DM, Bouzigues A, Swift IJ, Peakman G, Bocchetta M, Todd EG, Convery RS, Rowe JB, Borroni B, Galimberti D, Tiraboschi P, Masellis M, Finger E, van Swieten JC, Seelaar H, Jiskoot LC, Sorbi S, Butler CR, Graff C, Gerhard A, Langheinrich T, Laforce R, Sanchez-Valle R, de Mendonça A, Moreno F, Synofzik M, Vandenberghe R, Ducharme S, Le Ber I, Levin J, Danek A, Otto M, Pasquier F, Santana I, Kornak J, Boeve BF, Rosen HJ, Rohrer JD, Boxer AL, Frontotemporal Dementia Prevention Initiative (FPI) Investigators. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med. 2022 Oct;28(10):2194-2206. Epub 2022 Sep 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
AbbVie
This is a fantastic study in the sense that for the first time we now have data on reasonably large groups of patients with genetically defined forms of FTD and we can directly track changes on biomarkers and clinical end points over time. Long overdue!
The data suggest that GRN carriers have a more aggressive course, followed by MAPT and then C9ORF carriers in both clinical and biomarker readouts. While the differences seem to be lost when looking at a global measure (CDR+NACC), the differential rates of progression for more sensitive items could easily enable more adaptive designs.
Assuming that one runs a trial in which all three forms of FTD are enrolled, then knowing about these differential rates would enable more efficient interim analyses and more efficient utilization of subjects with very rare disorders, e.g., tailoring the numbers of subjects to enroll and their duration of treatment, etc.
I think this is a beautiful example of how coordinated, highly collaborative research efforts are necessary to move the needle. The tempo and type of biomarker change varies by actual etiology—e.g., distinct genetic forms have distinct early presentations and trajectories in terms of biomarkers. This fits with the framing of neurodegenerative disorders, such as AD, FTD, PD, etc., more generally as syndromes, rather than discrete diseases. This is similar to cancer, where the underlying biological drivers, such as specific mutations, manifest distinct clinical challenges and treatments.
VU University Medical Center
This study provides relevant and reliable information about the trajectory of changes of known biomarkers for familial FTD, namely atrophy and NfL levels, before and after latent disease age. It informs trial design in terms of numbers to include per FTD mutations in C9ORF72, GRN, and MAPT. Compared to GRN and MAPT mutation carriers, about twice as many C9ORF72 and MAPT mutation carriers would have to be included in two-year trials if the endpoint is a clinical one, namely CDR+ NACC-FTD-SB. The numbers are even more divergent if MRI would be chosen as primary endpoint.
Other outcomes are remarkable: the consistency of the results between the centers, located in North America and Europe; the heterogeneity in results and the consequent lack of thresholds for inclusion, which show that it is hard to use MRI and NfL for inclusion at the individual levels. That the NfL increases largely coincided with the earliest clinic symptoms, and often in parallel with increases in atrophy in medial, temporal, or frontal lobes, corroborates the order of changes in, for example, AD, and is a consequence of NfL being an axonal damage marker, lying downstream of specific neuronal disruptions. The thalamic atrophy observed in a very early stage in C9ORF72 mutation carriers implies that the earliest biological alterations can be measured quite well. It is useful for trial design to understand the prognostic value of these regional atrophy changes.
Large, multicenter collaborations, similar to this one by Stafforoni et al., are needed, and they will enable the precise mapping of f-FTD mutation-specific changes. Our efforts should be directed toward the identification of low-invasive biomarkers for the earliest changes, and toward inclusion thresholds to further support trial inclusion.
Make a Comment
To make a comment you must login or register.