Does ‘Runaway Aggregation’ Spell the End for Neurons?
Quick Links
How long can someone bail water before the boat tips over? That depends on two opposing factors: the rush of water, and the fitness of the bailer. A similar push-and-pull may take place in neurodegenerative proteinopathies, where disease hinges on a balance between the production of aggregates, and how efficiently cells can gid rid of them. This is the upshot of a preprint posted October 30 on bioRxiv.
- Mathematical modeling of the dynamics between protein accumulation and removal defines a pathological tipping point.
- Beyond that point, “runaway aggregation” ensues, transforming a healthy cell into one laden with tangles—or Lewy bodies.
- Preventing aggregation works better than speeding clearance.
Combining high-resolution imaging with mathematical modeling and cellular assays, researchers led by Georg Meisl and David Klenerman of the University of Cambridge, U.K., and Randal Halfmann of the Stowers Institute for Medical Research in Kansas City, Missouri, defined a cellular tipping point, beyond which aggregation becomes “runaway.” At that point, a cell transitions from a healthy state to a pathological one.
Brain samples from people with Alzheimer’s or Parkinson’s disease harbored a high proportion of neurons that had crossed this point of no return, exemplified by an overload of tau tangles or α-synuclein aggregates, respectively. The findings support the idea that therapies that prevent cells from reaching the tipping point stand a better chance of success than those aiming to restore order to cells that have already crossed to the other side. Which side of the equation to target—aggregate production versus removal—could come down to disease stage and subtype, the authors contend.
To some scientists Alzforum contacted, the findings largely bundle years of previous observations into mathematical packaging. Others viewed the study as a fundamental contribution to our understanding of neurodegenerative disease.
“This remarkable paper sets out to derive a mathematical model that can explain why tau and α-synuclein aggregates, which occur in normal aging, transition to become pathological in Alzheimer’s and Parkinson’s diseases,” wrote Scott Small of Columbia University in New York. “The manuscript compellingly concludes that the shift toward pathogenic aggregate proliferation is primarily driven by degradation defects” (comment below).
Meisl and Klenerman previously wielded computational tools to come to the provocative conclusion that the doubling of the number of existing tau seeds within cells, rather than an uptick in their propagation between cells, is the main driver of tauopathy progression (Nov 2021 news). Their latest mathematical venture attempts to bridge the gap between observations made at the molecular level—namely, protein aggregation—and those made at the brain-wide scale, i.e., proteopathic progression and neurodegeneration. According to the authors, the seeded propagation idea fails to account for low levels of aggregates in healthy cells in unaffected brain regions, or in people without neurological disease. Furthermore, aging and faltering proteostasis—not the production of a single instigating aggregate—are the major risk factors for neurodegenerative disease.
“These observations imply that organisms can tolerate, or even limit, aggregation for extended periods, pointing to additional control layers, such as active cellular mechanisms, that continually counteract the ongoing accumulation,” the authors wrote. “An alternative hypothesis then emerges, that, in addition to aggregates self-replicating, an imbalance of aggregate production and removal is the crucial step in the emergence of disease.”
To test this idea, first author Matthew Cotton and colleagues started by looking at the distribution of α-synuclein aggregates in brain samples from people with PD and from non-diseased controls. Using confocal microscopy, they spotted intraneuronal clumps of α-synuclein in both. While healthy brains had mainly small aggregates, in PD brains the aggregates had exploded in size within some cells, suggesting the removal machinery had been overwhelmed (image below).
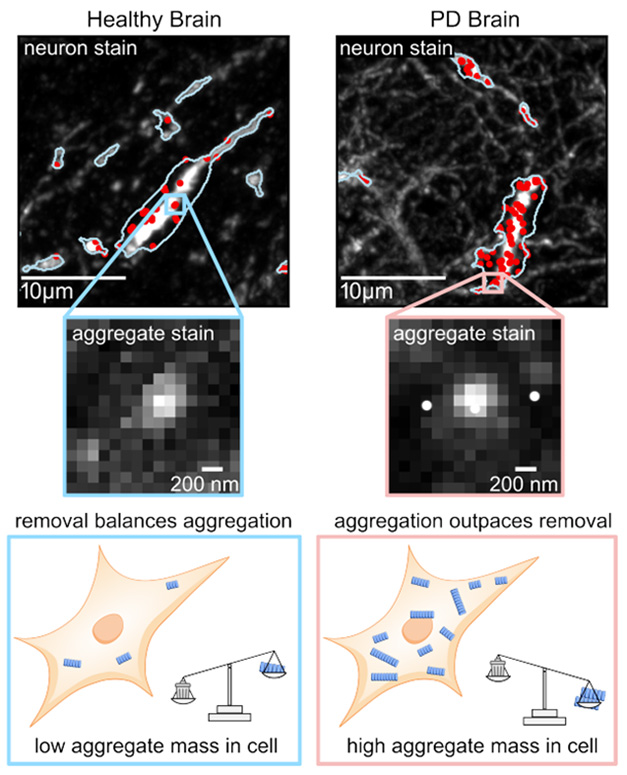
Tip the Scales. Immunofluorescence imaging (top) detects α-synuclein aggregates within neurons in healthy (left) and PD (right) brains, with larger aggregates seen in PD. Those occur when aggregation outpaces removal (schematic, bottom). Cells that have passed this tipping point are more abundant in the PD brain. [Courtesy of Cotton et al., 2025, bioRxiv.]
They reasoned that in cells that had reached this tipping point, larger aggregates would grow and persist for longer time periods, resulting in a quantifiable shift in aggregate size distribution. To measure this, Cotton and colleagues deployed single-aggregate imaging on brain homogenates. In brief, this high-resolution technique quantifies the length of individual aggregates extracted from a brain sample. In keeping with their hypothesis, they found a hefty shift in the size distribution within PD brain samples. Similarly, AD brain samples amassed larger aggregates of phosphorylated tau than seen in control brains (image below).

Overwhelmed. The size distribution for α-synuclein (left) and tau (right) shifted toward longer aggregates in people with PD or AD (red) relative to healthy controls (blue). [Courtesy of Cotton et al., 2025, bioRxiv.]
The authors developed a mathematical model to decipher these dynamics. Applying it to their aggregate size distribution data, they found that in PD brains, 82 percent of α-synuclein aggregates occurred in cells that had moved beyond their tipping point. In those cells, aggregate removal had cratered by 61 percent relative to that in healthy cells. In AD brain samples, a similar phenomenon emerged, with 89 percent of intracellular tau aggregates found in cells beyond their tipping point, and aggregate removal slowed by 62 percent.
Further mathematical models predicted that, in healthy cells, aggregates are continually produced and dealt with via degradation mechanisms. However, once aggregate accumulation outpaces removal, “runaway aggregation” drives the cell into a pathological state. “Once cells have made that transition, it is significantly more difficult to move them back to their healthy state than it is to prevent the transition in the first place,” the authors wrote.
To test this idea experimentally, the authors devised a high-throughput cellular assay, in which they transfected cells with an increasing number of plasmids carrying a copy of Aβ42, along with FRET fluorophores to measure aggregation. Two populations emerged from these mixed cultures: one with low numbers of aggregates, and the other with an aggregate overload. The switch between these two states was sharp, not gradual. It occurred at a critical number of plasmids, i.e., concentration of Aβ monomers. What’s more, the scientists were able to shift this tipping point by co-expressing an Aβ aggregation inhibitor along with Aβ.
The results not only support the tipping-point hypothesis, but also suggest that in situations where monomers abound, such as in carriers of an α-synuclein triplication, or in people with Down’s syndrome, excess monomer pushes cells across the line.
“From these data, their model predicts a ‘sudden transition’ at critical monomer concentrations determined by substrate concentration. This is consistent with our biofluid analyses of Aβ changes in AD natural history,” commented Colin Masters of the University of Melbourne in Australia.
The scientists explored what else might tip the balance toward runaway aggregation in the presence of tau seeds. Using both tau biosensor cell lines and hippocampal slice cultures from P301S-tau mice, they found that the closer a cell gets to its tipping point, the fewer tau seeds are required to push it over the edge. For example, if a cell overexpresses tau monomers, or is in a compromised state of oxidative stress, fewer tau seeds are needed to spark rampant aggregation.
The findings point to feedback mechanisms, whereby a growing burden of aggregates—perhaps derived from different proteins such as Aβ, tau, α-synuclein, or TDP-43—gum up removal systems. “A limited capacity for removal naturally explains the emergence of co-aggregates and co-pathologies,” the authors wrote. “Aggregates of one protein slow generic removal processes, thus moving other proteins closer to their respective tipping points.” Mixed pathologies, anyone?
Small noted another variation of this type of vicious cycle, whereby tau aggregates sequester retromer proteins. They facilitate the recycling of endosomal proteins to the cell surface and to the trans-Golgi network, and ultimately to the lysosome (Morderer et al., 2025; Young et al., 2023). In so doing, tau aggregates put a wrench in their own removal, and potentially that of other proteins, he suggested.
Meisl told Alzforum that these feedback mechanisms potentially explain the connection between Aβ and tau pathologies in AD. Perhaps buildup of extracellular Aβ stresses neurons and hobbles their lysosomal function, inching them closer to the tipping point for runaway aggregation of tau.
In that vein, Eric McDade of Washington University in St. Louis, noted that when neurons are stressed—whether by Aβ aggregates or other factors—they crank up phosphorylation of tau, thus inadvertently increasing the concentration of tau seeds in the vicinity. Other cell types, such as microglia and astrocytes, are also involved in clearance mechanisms, and their role in tipping the balance toward aggregation versus removal should be investigated, McDade said.
McDade wondered how protective mutations, such as the ApoE3-Christchurch variant, might tip the scales. The ApoE3-Ch variant staved off dementia in a pathogenic Paisa mutation carrier for decades. Her brain was riddled with amyloid plaques, yet had few tangles on PET (Sep 2022 news). Similarly, a participant in the DIAN cohort has so far managed to escape his fate, possibly due to copious heat shock proteins coursing through his cerebrospinal fluid (Feb 2025 news). Both mechanisms are thought to work by improving removal of tau aggregates, either at the level of glia in the case of ApoE3-Ch, or by way of chaperones, in the latter case (Dec 2023 news).
“They have stress in the system, in terms of amyloid plaques, but their improved removal mechanisms may lower the tipping point for tau,” McDade proposed.
Masters also noted that the effect of ApoE haplotype—which dictates the age at onset of detectable Aβ aggregation, rather than the subsequent pace of progression—could be considered in future work. “Surely this must be the strongest evidence available for the seeding effect being the dominant effector of onset and progression (but not severity) of late-onset (sporadic) AD?” Masters wrote.
Ralph Nixon of New York University, Orangeburg, interpreted the findings in a different light. He views the passage of the aggregation tipping point as a canary in a coalmine, reflecting a failure of the lysosomal system. When proteostasis falters, due to a convergence between genetic risk factors and aging, aggregation-prone proteins such as α-synuclein and tau will be the first visible sign of this incipient dysfunction. Once lysosomes fail, autophagy fails, and the cell no longer regulates energy production or nutrient homeostasis, or carries out other essential functions. This, rather than the aggregation of any one protein, is what kills the cell, Nixon posits.
The findings have implications for therapeutic intervention. Not only do they buttress the notion that earlier is better, they also hint at which types of therapies might work best at different stages of disease. “This validates the idea that aggregate removal strategies are important for therapeutic design in neurodegeneration, but so are aggregation inhibitors,” commented Jessica Young of the University of Washington in Seattle. “While interesting, the model presented here is still a simple system,” she added, noting that future studies could consider how different biological parameters, such as lysosome biogenesis and turnover, age, and cell type, come into play.—Jessica Shugart
References
News Citations
- Doubling of Tau Seeds, Not Spread, Sets Pace of Tauopathy in Alzheimer's
- In Brain With Christchurch Mutation, More ApoE3 Means Fewer Tangles
- Hot News: DIAN Participant Defies Presenilin 2 Mutation
- APOE Christchurch Variant Tames Tangles and Gliosis in Mice
Paper Citations
- Morderer D, Wren MC, Liu F, Kouri N, Maistrenko A, Khalil B, Pobitzer N, Salemi MR, Phinney BS, Bu G, Zhao N, Dickson DW, Murray ME, Rossoll W. Probe-dependent Proximity Profiling (ProPPr) Uncovers Similarities and Differences in Phospho-Tau-Associated Proteomes Between Tauopathies. Mol Neurodegener. 2025 Mar 13;20(1):32. PubMed.
- Young JE, Holstege H, Andersen OM, Petsko GA, Small SA. On the causal role of retromer-dependent endosomal recycling in Alzheimer's disease. Nat Cell Biol. 2023 Oct;25(10):1394-1397. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Cotton MW, Venkatesan S, Beckwith JS, Böken D, Xu CK, Fertan E, Breiter JC, Berkowicz LE, Salazar LS, Von Schulze A, Andrzejewska EA, Brock EE, Han HL, Schneider MM, Sahtoe DD, Baker D, Rowe JB, Goriely A, McEwan WA, Knowles TP, Lee SF, Halfmann R, Klenerman D, Meisl G. Neurodegeneration emerges at a cellular tipping point between aggregate accumulation and removal. bioRxiv. 2025 Oct 30; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Columbia University
Most of us have at least intuited that nonlinear dynamics are embedded within neurodegenerative disorders. Most of us are not mathematicians. This remarkable paper sets out to derive a mathematical model that can explain why tau and α-synuclein aggregates, which occur in normal aging, transition to become pathological in Alzheimer’s and Parkinson’s diseases. Not a mere theoretical model or rehashing of prior findings, the manuscript is distinguished by: A) generating a model based on new microscopic tools capable of quantifying aggregate size in postmortem neurons; and B) testing and validating inferences via experimental manipulations. Besides nontrivial descriptions of how neuronal aggregates are formed, it is the mechanistic and therapeutic insights derived from the validated model that represents the manuscript’s greatest impact.
The manuscript compellingly concludes that the shift toward pathogenic aggregate proliferation is primarily driven by degradation defects. By slowing removal rates, a nonlinear, “runaway” proliferation will occur, racing toward a tipping point beyond which pathological aggregation accumulation is inevitable and unavoidable. Strikingly, the defective removal rates are estimated to be nearly identical in AD and PD, suggesting a common degradation mechanism.
As the authors emphasize, the runaway acceleration might occur by overloading the neuron’s degradation machinery or even by a vicious cycle that might exist between degradation defects and aggregate accumulation. More important for patients, the manuscript’s findings justify its therapeutic conclusions. Particularly relevant for therapeutics is the pointed conclusion that “aggregate inhibitors” are likely required to restore neuronal health in incipient disease, during which time they are likely to have greater clinical efficacy than aggregate-clearing interventions.
Since the authors invoke the lysosome-autophagosome as one possible defective degradation mechanism, their conclusions might relate to a highly pathogenic biological pathway that many of us focus on—retromer-dependent endosomal recycling (Young et al., 2023).
Recent multi-omic studies by Pete Cullen’s lab (Daly et al., 2023) and hypothesis-driven studies by others have established how critical this pathway is for lysosome-autophagosome health. This explains why the pathway regulates tau aggregation, by influencing both their production and their degradative removal. With potential relevance to this manuscript, these observations were incorporated into the conclusions of a recently published study. In it, tau aggregates were found to sequester retromer proteins, and a potential vicious cycle between tau aggregation and pathway dysfunction was suggested (Morderer et al., 2025).
More than its specific results, this manuscript introduces and validates a framework for clarifying mechanisms that relate to proteinopathies across neurodegenerative disorders. Indeed, the authors suggest that the derived conclusions regarding tau and α-synuclein might represent a “universal feature.” It is now known that most of the common neurodegenerative disorders are characterized by a mixture of multiple protein aggregates—not just formed around tau and α-synuclein but also TDP-43 and TMEM106b. Future studies can rely on this framework to test whether the dynamics of aggregate proliferation described in this manuscript generalize to these other proteinopathies, and whether they are driven by similar degradation mechanisms.
References:
Daly JL, Danson CM, Lewis PA, Zhao L, Riccardo S, Di Filippo L, Cacchiarelli D, Lee D, Cross SJ, Heesom KJ, Xiong WC, Ballabio A, Edgar JR, Cullen PJ. Multi-omic approach characterises the neuroprotective role of retromer in regulating lysosomal health. Nat Commun. 2023 May 29;14(1):3086. PubMed.
Morderer D, Wren MC, Liu F, Kouri N, Maistrenko A, Khalil B, Pobitzer N, Salemi MR, Phinney BS, Bu G, Zhao N, Dickson DW, Murray ME, Rossoll W. Probe-dependent Proximity Profiling (ProPPr) Uncovers Similarities and Differences in Phospho-Tau-Associated Proteomes Between Tauopathies. Mol Neurodegener. 2025 Mar 13;20(1):32. PubMed.
Young JE, Holstege H, Andersen OM, Petsko GA, Small SA. On the causal role of retromer-dependent endosomal recycling in Alzheimer's disease. Nat Cell Biol. 2023 Oct;25(10):1394-1397. PubMed.
University of Melbourne
Cotton and colleagues from the Department of Chemistry at University of Cambridge are evaluating the kinetics of aggregating proteins/peptides (Aβ, α-synuclein, and tau) in their balance between accumulation and clearance leading to a tipping point beyond which irreversible “pathological” changes occur. They use super-resolution microscopy of human brain sections, in vitro cell-based and organotypic assays together with mathematical models. They try to interpret their data in relation to disease onset, progression, and severity.
There appears to be some lack of definition of “healthy” human brain samples, which have small numbers of α-syn and tau aggregates which would normally be considered part of the preclinical spectrum of AD and PD. More importantly, the fundamental question that is not addressed relates to the, as yet, unanswered conundrum of how does extracellular Aβ drive intracellular α-syn and tau aggregation? There is increasing evidence of Aβ/α-syn/tau interactions (e.g., Röntgen et al., 2025) confirmed by long-standing clinico-pathological studies of DLB and the effect of immunotherapies targeting Aβ, which lower tau/p-tau levels.
The imbalance between aggregate production and clearance is the focus of this manuscript; however, the kinetics of their in vitro cell/tissue assays are at variance with the time course of human AD and PD. Nevertheless, the concept of a “tipping point” could be very relevant, and is being confirmed with current studies of biofluid and molecular imaging of Aβ changes over the natural history of AD. A key point of their study is that the balance between aggregate growth and removal is determined specifically when fibrillar “aggregates are long enough for their growth rates to no longer be size-dependent.”
Surprisingly, the authors calculate that the average length of an α-syn and tau aggregate in a “healthy” brain is fewer than 1,000 monomers, and that α-syn in a PD brain is ≅ 2,000 monomers and tau in an AD brain is ≅ 3,000 monomers. They also calculate that in PD and AD the relative removal rate of both α-syn and tau aggregates is reduced by 60 percent. From these data, their model predicts a “sudden transition” at critical monomer concentrations determined by substrate concentration. This is consistent with our biofluid analyses of Aβ changes in AD natural history.
Can seeding be triggered by a single aggregate? Probably not. Using organotypic hippocampal slice cultures and other cell-based assays, the authors conclude that multiple seeds are required to trigger aggregation, and also concede that other cellular micro-environs are likely to be critical. They quote the effect of cholesterol-lowering agents on cytosolic access of tau aggregates, which lower the numbers of aggregates required for critical seeding. Strangely, they do not refer to the mechanism of action of ApoE, the major genetic risk for AD, where evidence is now building for an effect of the lipidation state of the 2, 3, and 4 ApoE haplotypes.
And this leads to another fundamental question which could have been addressed in their cell/tissue-based assays: ApoE haplotype strictly determines the age of onset of AD (ε4 carriage about seven years earlier than ε4 non-carriage), and yet the slopes of Aβ accumulation are independent of ε4 carriage. Surely this must be the strongest evidence available for the seeding effect being the dominant effector of onset and progression (but not severity) of late-onset (sporadic) AD?
References:
Röntgen A, Toprakcioglu Z, Vendruscolo M. Aβ42 promotes the aggregation of α-synuclein splice isoforms via heterogeneous nucleation. FEBS Lett. 2025 Oct;599(19):2750-2767. Epub 2025 Jul 26 PubMed.
University of Washington
This interesting work by Cotton and colleagues uses mathematical models to predict protein aggregation. The models show that pathological protein aggregation occurs when either too much protein is being produced, or is not being removed fast enough. This is validated in cellular systems. Altogether, this validates the idea that aggregate removal strategies are important for therapeutic design in neurodegeneration, but so are aggregation inhibitors.
The model presented here is still a simple system. It would be interesting to incorporate how normal cellular functions, such as lysosome biogenesis and turnover, that regulate protein degradation could be incorporated, especially as these cellular processes may differ between different cell types and with age.
Make a Comment
To make a comment you must login or register.