Droplets in the Bucket? Introducing en Masse Single-Synapse RNA-Seq
Quick Links
Synaptic changes lie at the nexus of many neurodegenerative diseases, underpinning cognitive decline. However, it has been hard to get a handle on exactly how these tiny structures change in disease, because scientists have had no way to examine expression changes in single synapses at scale. In the January 16 Nature Biotechnology, researchers led by David Weitz at Harvard University and Chenghang Zong at Baylor College of Medicine in Houston present a high-throughput way to profile the RNA composition of thousands of individual synapses. Going by the mouthful Multiple-Annealing-and-Tailing-based Quantitative scRNA-Seq in Droplets, or MATQ-Drop, the method isolates nuclei or synaptosomes from brain, encases each in an oil droplet that adds a unique barcode, and then mixes the labeled droplets for one large-scale RNA-Seq.
- New method permits high-throughput RNA-Seq of single synapses.
- It identifies distinct synaptic profiles in mouse and in human hippocampus.
- With amyloidosis, inflammatory and complement transcripts shoot up.
With this approach, the authors identified 12 distinct synaptic profiles in wild-type mouse hippocampus, and six in postmortem human hippocampus, confirming that synapses within a brain region vary at the RNA level. The scientists think they might uncover more human profiles if the postmortem interval were shorter. In the 5XFAD mouse model of amyloidosis, the 12 synaptic subtypes expressed higher levels of inflammatory factors, especially complement, than did synapses from control mice.
“This approach opens a whole new avenue of biology for neuroscientists to look into synapses at the individual level,” Zong told Alzforum.
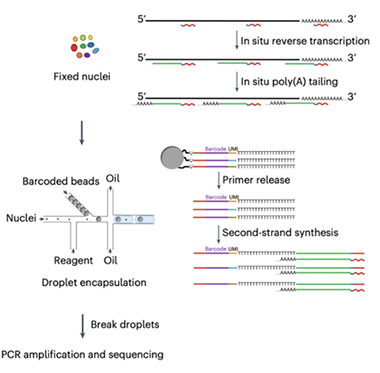
Two-Step RNA-Seq. Used with either isolated nuclei (pictured) or synaptosomes, MATQ-Drop starts with internal primers to reverse-transcribe mature, immature, and noncoding RNA; in the second step, beads add a unique barcode to individual nuclei or synaptosomes in separate oil droplets. [Courtesy of Niu et al., Science Advances/AAAS.]
Others agreed. “This technology may help us better understand the diversity of synapses in the brain in health, and how they change in disease,” Robert Vassar at Northwestern University, Chicago, wrote to Alzforum. Ulrich Hengst at Columbia University, New York, noted differences in transcript splicing among synapses. “The paper provides several intriguing starting points for future research,” Hengst said (full comments below).
Why analyze RNA in synapses? Previous work has found that neurons shuttle transcripts to both pre- and post-synapses where regulated local translation allows for rapid change in protein composition in response to stimuli (May 2019 news). Those studies examined synapses in bulk, however, making it impossible to discern whether there were subtypes that might have specific agendas. To achieve single-synapse resolution, the authors combined two existing methods, the MATQ-Seq technology developed by Zong, and the droplet sequencing method developed by Weitz and colleagues at Harvard (Sheng et al., 2017; Sheng and Zong, 2019; Macosko et al., 2015).
MATQ uses primers that bind at several sites along a transcript, rather than only at the poly-A tail. This allows for reverse transcription of total RNA, including immature, unspliced, and long noncoding RNAs (lncRNA), as well as mature transcripts. LncRNAs serve as scaffolds for transcripts and help regulate gene expression. Applying the MATQ technique to synapses was challenging, however, because these structures are open rather than membrane-bound like a nucleus, which means RNA leaks out. To prevent this, first author Muchun Niu fixed synaptosomes immediately after isolating them, locking RNA in place before reverse transcription. Then the authors used fluorescence-activated sorting to separate synaptosomes, merging each one with a drop of oil containing bar-coded oligonucleotides. Enzymes in the oil added the oligos to the cDNA strands from the reverse transcription step, labeling each synapse’s transcriptome before large-scale sequencing.
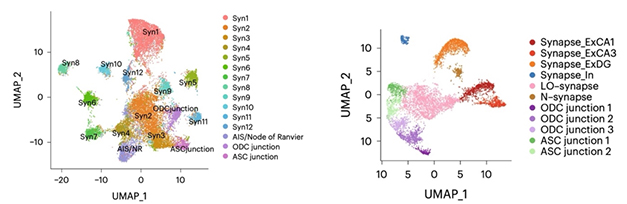
Mouse versus Human. In mouse hippocampus, MATQ-Drop found 12 distinct synaptic signatures (left), plus neuron-oligodendrocyte (ODC), neuron-astrocyte (ASC), and axon initial segment (AIS) signatures. In human hippocampus, six synaptic, three ODC, and two ASC junction signatures were captured (right). [Courtesy of Niu et al., Nature Biotech.]
Applying this method to mouse hippocampus, the authors identified 12 distinct synaptic profiles, or “synaptomes.” They also found axon initial segments, as well as what the authors called neuron-astrocyte and neuron-oligodendrocyte “junctions,” i.e., places where these cell types meet. The junctions appeared because the technique for isolating synaptosomes picked up other subcellular structures as well.
Two of the synaptic profiles contained transcripts for known presynaptic markers such as PCLO and BSN, and one contained postsynaptic markers, such as SHANK1, but the nature of the other subtypes was less clear. Notably, each synaptic subtype had a distinct signature of unspliced mRNA and of lncRNA as well. Comparing these synaptic profiles to single-nuclei RNA-Seq from the same hippocampal samples, the authors found that while there was some correspondence between synaptic and neuronal subtype transcriptomes, they did not match up cleanly. This implies that some synaptic subtypes are common to several types of neuron.
In postmortem hippocampus and prefrontal cortex samples from two human brains, the researchers found fewer RNA profiles, but these matched up better with neuron transcriptome subtypes. Three synaptomes came from excitatory neurons, two from inhibitory, and one seemed to mark immature synapses. The differences between mouse and human samples might be partly due to the long postmortem interval for the human samples, allowing RNA to decay, the authors speculated. As in mouse, they detected neuron-astrocyte and neuron-oligodendrocyte “junctions,” as well as subtype-specific lncRNAs.
What happens in AD? In 20,000 hippocampal synaptosomes isolated from two 5XFAD and two wild-type mice, transcripts for inflammatory genes, particularly complement proteins, shot up. C1q quadrupled compared to wild-type synapses. Complement proteins, especially C1q, have been implicated in synapse loss in AD (Nov 2015 conference news; Jun 2022 news). The authors found other changes as well, such as more myelination transcripts, and a switch in the type of calcium sensor from Camk2d to Camk2a, potentially affecting synaptic plasticity (Zalcman et al., 2018). Crucially, these synaptic changes were not reflected in single-nuclei RNA-Seq data from the same samples, indicating that synaptic profiling captures unique data.
Finally, the authors compared MATQ-Drop to a commercial platform for single-nuclei RNA-Seq, the 10x Genomics Chromium. When both methods were applied to nuclei isolated from mouse hippocampus, MATQ-Drop was more sensitive, detecting a third more unique neuronal transcripts and more than twice as many glial transcripts as did 10x Genomics Chromium. “Because of our total RNA capture, we have much more efficient gene detection compared to the latest commercial platforms,” Niu said.—Madolyn Bowman Rogers
References
Research Models Citations
News Citations
- Plasticity Hums With Protein Synthesis on Both Sides of Synapse
- Microglia Control Synapse Number in Multiple Disease States
- C1q Shows Promise as Therapeutic Target to Stop Synapse Loss
Paper Citations
- Sheng K, Cao W, Niu Y, Deng Q, Zong C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat Methods. 2017 Mar;14(3):267-270. Epub 2017 Jan 16 PubMed.
- Sheng K, Zong C. Single-Cell RNA-Seq by Multiple Annealing and Tailing-Based Quantitative Single-Cell RNA-Seq (MATQ-Seq). Methods Mol Biol. 2019;1979:57-71. PubMed.
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015 May 21;161(5):1202-1214. PubMed.
- Zalcman G, Federman N, Romano A. CaMKII Isoforms in Learning and Memory: Localization and Function. Front Mol Neurosci. 2018;11:445. Epub 2018 Dec 4 PubMed.
External Citations
Further Reading
News
- Single Synapse Mass Spec Snags CD47 as Alzheimer’s Resilience Factor
- Does Calcium Overload Mark Dendritic Spines for Destruction?
- Neuronal SRPX2 Spoils Microglial Appetite for Synapses
- Nixing Complement Protein Protects Neurons in Tauopathy Model
- Tau Silences, Aβ Inflames; Hitting Excitatory Synapses Hardest
Primary Papers
- Niu M, Cao W, Wang Y, Zhu Q, Luo J, Wang B, Zheng H, Weitz DA, Zong C. Droplet-based transcriptome profiling of individual synapses. Nat Biotechnol. 2023 Jan 16; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Northwestern University Feinberg School of Medicine
The approach is interesting and may provide insights into synaptosome characteristics that reflect synaptic function. Many mRNAs translated in the synaptic spine, such as neurotransmitter receptors, are important for post-synaptic function and regulation. This technology may help us better understand diversity of synapses in the brain in health, and how they change in disease.
Columbia University
As opposed to the currently available bulk-sequencing approaches, this MATQ-Drop method allows for profiling of the transcriptome at individual synapses. There are various types of synapses in the brain and, importantly, they are differently affected in neurodegenerative disorders. This new method may now allow us to discover synapse-type-specific changes in the local transcriptome in Alzheimer’s disease.
The strength of this method lies in the combination of single-synapse sequencing with single-nuclei sequencing, as the authors have done here. Additionally, because the method allows comparison of mature and nascent RNA, it will be useful to the elucidate how the local splicing landscape in individual synapse types is altered in conditions such as AD.
The paper is data-rich and provides several intriguing starting points for future research. For example, in AD, expression of three complement component genes were significantly changed in synapses, but not in nuclei, indicating that their transport and recruitment to synapses is upregulated. It will be interesting to find out what processes control the post-transcriptional regulation of these genes in the context of neurodegeneration. More generally, the data presented in his study emphasize the importance of changes to local translation in AD pathogenesis.
Ann Romney Center for Neurologic Diseases, Brigham and Women's Hospital and Harvard Medical School
I am very impressed with this new methodology for examining gene-expression changes at the synaptic level. This is a very clever technique and it does appear to get past some issues with transcriptomic analysis, including single-nucleus RNA-Seq, which could easily miss the synapses present on neurites.
I am not surprised to see both C3 and C1q involvement in the microglial synaptome. Both play a role in microglial-mediated synaptic pruning, as Beth Stevens, Dori Schaefer, Andrea Tenner, Susana Rosi, and my labs have demonstrated in the past. Overall, this looks like a big step forward in CNS transcriptomics, especially since synapses are so relevant to neurological aging and disease. I hope it will be available to the field at large in the near future.
The University of Tokyo
In this study, the authors have combined MATQ-Seq, a highly sensitive quantitative single-cell RNA-Seq technology they have developed, with droplet technology to enable quantitative RNA-Seq at high resolution. By using MATQ-Drop, they were able to analyze synaptosomes, which are enriched in RNAs distinct from those in the cytoplasm, at the single-compartment level. The authors successfully detected transcriptome differences at the synaptic level in mice, humans, and in a mouse model of Alzheimer's disease.
This is a very important technique and has clarified RNA abnormalities in the specialized compartment of the synapse. This technique is also interesting because it has the potential to detect transcriptome abnormalities in various compartments and organelles in the cell if their biochemical isolation is possible. For example, it has the potential to reveal new "intracellular transcriptome diversity" in the cytoplasm, which was believed to be a single environment before the biology of by liquid-liquid phase separation was discovered.
The finding, in mice, of neuron-oligodendrocyte junctions might be further interesting because, in addition to microglia, oligodendrocyte precursor cells (OPCs) prune synapses in the mouse visual cortex (Auguste et al., 2022).
References:
Auguste YS, Ferro A, Kahng JA, Xavier AM, Dixon JR, Vrudhula U, Nichitiu AS, Rosado D, Wee TL, Pedmale UV, Cheadle L. Oligodendrocyte precursor cells engulf synapses during circuit remodeling in mice. Nat Neurosci. 2022 Oct;25(10):1273-1278. Epub 2022 Sep 28 PubMed.
Make a Comment
To make a comment you must login or register.