“Frustrated Oligomers” Slow Aggregation of Aβ42
Quick Links
When it comes to Aβ peptides, short may very well be sweet. In the February 7 Chemical Science online, researchers led by Sara Linse, University of Lund, Sweden, report that Aβ37, Aβ38, and Aβ40 all block the formation of Aβ42 aggregates, and that combinations of the shorter peptides work better than any of them alone. Though this work was conducted in vitro, scientists believe that similar dynamics likely play out in the human brain, where the shorter versions of Aβ can constitute the lion’s share. The findings help explain the toxicity of mutations that cause familial Alzheimer’s disease, many of which reduce levels of Aβ37/Aβ38, and may buoy efforts to find γ-secretase modulators that favor production of shorter peptides.
- Aβ37 and Aβ38 form fibrils slowly.
- These short peptides slow formation of Aβ42 and Aβ40 fibrils.
- Mixtures of all four peptides retard fibrillogenesis even more.
“The results here clearly suggest that consideration of Aβ species outside of the heavily studied Aβ40 and 42 fragments may be necessary to comprehensively understand an individual’s level of β-amyloid pathology and how it relates to risk of AD-related cognitive decline,” wrote Jasmeer Chhatwal, Lei Liu, and Dennis Selkoe, Brigham and Women’s Hospital, Boston (comment below).
Researchers have previously reported that the smaller Aβ isoforms can dampen toxicity and growth of Aβ42 fibrils in vivo and in vitro (Moore et al., 2018; Quartey et al., 2021). The new analysis is more comprehensive. Using thioflavin T binding and cryo-transmission electron microscopy, first author Gabriel Braun and colleagues not only tested how Aβ37, Aβ38, and Aβ40 affect growth of Aβ42 fibrils, but how combinations of all four dictate fibril dynamics, both as monomers and as fibrillogenic seeds.
First, Braun tested if Aβ37 and Aβ38 monomers form fibrils by themselves. They did, albeit much more slowly than did Aβ40 or Aβ42 monomers. While the latter aggregated in less than an hour, and Aβ40 in a couple of hours, the shorter peptides took days, although they did so in much the same way eventually, namely via secondary nucleation off existing fibrils (May 2013 news ).
Next the authors asked what would happen if they mixed different monomers. They titrated solutions of Aβ37 or Aβ38 with increasing concentrations of the longer peptides. Seen by thioflavin T fluorescence, Aβ40 decreased the lag time before aggregation of the shorter peptides took off, but did not change the shape of the sigmoidal curve. This is a kinetic hint that Aβ40 can form hybrid fibrils with the shorter isoforms.
Cryo-TEM supported this. Fibrils formed when Aβ40 was mixed with either of the shorter isoforms looked different than those formed by pure Aβ40, or by pure Aβ37 or Aβ38. The findings support the idea that Aβ40 co-aggregates with either Aβ37 or Aβ38 to form fibrils that have a distinct morphology from those formed by pure peptides.
Aβ42 decreased the lag time of Aβ37 and Aβ38 aggregation as well but, in this combination, the typical sigmoidal increase in fluorescence over time showed up as a doublet, meaning each monomer formed its own fibril in its own time (see image below). The scientists previously reported that mixtures of Aβ40 and Aβ42 behave similarly (Cukalevski et al.,2015).

Singlets, Doublets. During in vitro fibrillization assays, Aβ40 accelerates the aggregation of Aβ37 (top), as seen by a delay in thioflavin T fluorescence (y axes). Aβ42 also accelerated Aβ37 fibrillization (bottom), but in this case the typical sigmoidal increase in fluorescence was a doublet, suggesting that the two peptides form distinct fibrils in their own time. [Courtesy of Sara Linse.]
If the longer, more fibrillogenic isoforms can accelerate aggregation of the short forms, can the short form slow aggregation of Aβ40 and Aβ42? Indeed, Braun found that monomers of Aβ37 and Aβ38 delayed aggregation of the longer peptides in a concentration-dependent manner.
Similar kinetics dictated cross-seeding experiments. When the authors added pre-aggregated fibrils of Aβ40 or Aβ42 to monomers of Aβ37 or Aβ38, fibrillization of the shorter forms sped up. In fact, such seeds of any isoform catalyzed fibrillization of Aβ37 and Aβ38. Self-seeding was the most potent. The authors believe that the fibrils provide a surface for the primary nucleation of new fibrils. Interestingly, Aβ42 seeds were less potent than Aβ42 monomers in accelerating Aβ37 fibrillization, indicating that monomeric interactions may play an important role in in vivo dynamics.
Would seeds from the shorter forms accelerate fibrillization of the longer? Here again, Aβ40 and Aβ42 behaved differently. Seeds of either Aβ37 or 38 accelerated Aβ40 fibrillization but not Aβ42 kinetics. This indicates that the inhibition of Aβ42 fibrillization by Aβ37 and Aβ38 seen in de-novo fibrillization experiments, i.e. without seeding, must have been due solely to Aβ37 and Aβ38 monomers. “This is especially relevant given that, at the point in time at which Aβ42 aggregates, both Aβ37 and Aβ38 will be in a primary monomeric state, due to the relatively slow aggregation of both peptides,” wrote the authors.
“It is pretty clear that these types of test tube aggregation studies really do provide good insight. They have a high predictive value for in vivo effects of Aβ aggregation kinetics,” wrote Todd Golde, University of Florida, Gainesville, to Alzforum. But Golde thinks there is more going on in the brain. “Aβ prior to deposition is present at low nM levels and is likely bound to many other proteins, so how does Aβ42 ever nucleate in the brain?” he asked (comment below).
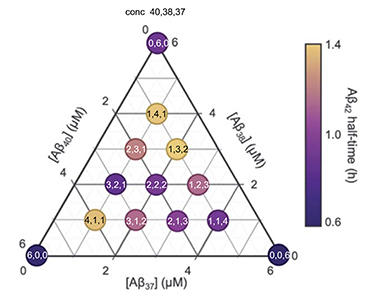
Triangulation. Combinations of Aβ37, Aβ38, and Aβ40 concentrations profoundly affect the half-life of monomeric Aβ42, which was longest when Aβ38 or Aβ40 levels were high. [Courtesy of Sara Linse, Lund University.]
Finally, what happens in the brain when there are complex mixtures of these monomers and fibrils? The authors began to look at this using three-peptide monomeric mixtures. In a nutshell, they found two distinct fibrillogenic process occurring in Aβ37/Aβ40/Aβ42 and Aβ38/Aβ40/Aβ42 mixtures. A faster aggregate formed that primarily comprised Aβ42, while fibrils consisting of the other peptides formed later. Mixtures of all four peptides behaved similarly.
Most notably from a physiological perspective, almost any mix of Aβ37/Aβ38/Aβ40 slowed the aggregation of Aβ42 (see image above). Further, while Aβ38 inhibited most strongly of the monomers, a 1:3:2 or 1:4:1 Aβ40/Aβ38/Aβ37 proved most effective. Linse thinks that the wider the variety of monomers in the mix, the more likely it becomes that “frustrated oligomers” form. The more heterogenous oligomers, the more difficult for them to assume the perfect repeated shapes that make up fibrils.
This findings comes on the heels of data from BioFinder, indicating that among people with early AD, those with the most Aβ38 decline most slowly (Jan 2022 news). “I find the paper from Sara Linse’s group very fascinating," wrote Oskar Hansson, also from Lund U. “It reinforces our recent findings in humans that higher levels of Aβ38 might be associated with a somewhat more benign disease course, by showing that in vitro aggregation of Aβ42 is slowed by Aβ38 and Aβ37.”
These recent studies may prompt scientists to take another stab at making safe and effective γ-secretase modulators. “To the extent that the presence of greater amounts of soluble Aβ37 and 38 relative to longer Aβ fragments is indicative of successful γ-secretase processivity, these results also bolster the case for using γ-secretase modulators to reduce Aβ plaque formation and lower the risk of AD-related cognitive decline,” Chhatwal and colleagues wrote.—Tom Fagan
References
News Citations
- Aβ Fibrils Drive Oligomer Formation, New Model Suggests
- Does More Aβ38 Mean Less Cognitive Decline in Alzheimer’s?
Paper Citations
- Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
- Quartey MO, Nyarko JN, Maley JM, Barnes JR, Bolanos MA, Heistad RM, Knudsen KJ, Pennington PR, Buttigieg J, De Carvalho CE, Leary SC, Parsons MP, Mousseau DD. The Aβ(1-38) peptide is a negative regulator of the Aβ(1-42) peptide implicated in Alzheimer disease progression. Sci Rep. 2021 Jan 11;11(1):431. PubMed.
- Cukalevski R, Yang X, Meisl G, Weininger U, Bernfur K, Frohm B, Knowles TP, Linse S. The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem Sci. 2015 Jul 1;6(7):4215-4233. Epub 2015 May 8 PubMed.
Further Reading
Primary Papers
- Braun GA, Dear AJ, Sanagavarapu K, Zetterberg H, Linse S. Amyloid-β peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-β 42 aggregation. Chem Sci. 2022 Feb 23;13(8):2423-2439. Epub 2022 Feb 7 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Massachusetts General Hospital / Harvard
Brigham and Women's Hospital
Co-Director, Brigham and Women's Hospital's Ann Romney Center for Neurologic Diseases
This is a thorough and thoughtful study that has potentially important implications for our understanding of Aβ aggregation in AD. The data clearly and convincingly demonstrate autocatalytic secondary nucleation as the predominant process by which Aβ37 and 38 form fibrils (similar to what has previously been shown for longer Aβ peptides). More importantly, the study's systematic approach to understanding the rates of fibril formation in 3- and 4-peptide mixtures demonstrates that cross-seeding of Aβ37 and 38 is readily observable using this biochemical approach. The complex interplay within and between Aβ alloforms includes both the inhibition of Aβ42 fibrillization in the presence of Aβ37 and 38 and promotion of Aβ37 and 38 aggregation in the presence Aβ42.
The authors also demonstrate that the cross-seeding of Aβ fragments leads to a variety of ultrastructurally distinct fibrils. This finding that raises the possibility that the relative proportions of Aβ alloforms in vivo may impact the fine ultrastructure of the β-pleated sheets produced from these fibrils. In turn, differences in β-pleated sheet ultrastructure based on cross-seeding could potentially impact the detectability of β-amyloid deposits by PET ligands and have important implications for the development and testing of AD therapeutics.
Importantly, however, it remains an open question as to whether the cross-seeding seen here in a pure biochemical preparation reflects what occurs in vivo, particularly as the concentrations of Aβ fragments used here are, in some cases, much higher than what is observed under physiologic conditions.
Nonetheless, the results here clearly suggest that consideration of Aβ species outside of the heavily studied Aβ40 and 42 fragments may be necessary to comprehensively understand an individual’s level of β-amyloid pathology and how it relates to risk of AD-related cognitive decline.
The observation that shorter Aβ fragments may inhibit aggregation of longer Aβ fragments (especially Aβ42) fits well with recent, intriguing work from Biofinder and ADNI that suggests having higher levels of shorter Aβ fragments, especially Aβ38, may be associated with lower rates of AD-related cognitive decline (Cullen et al., 2021).
To the extent that the presence of greater amounts of soluble Aβ37 and 38 relative to longer Aβ fragments is indicative of successful γ-secretase processivity, these results also bolster the case for using γ-secretase modulators to reduce β-amyloid plaque formation and lower the risk of AD-related cognitive decline.
References:
Cullen N, Janelidze S, Palmqvist S, Stomrud E, Mattsson-Carlgren N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative. Association of CSF Aβ38 Levels With Risk of Alzheimer Disease-Related Decline. Neurology. 2022 Mar 1;98(9):e958-e967. Epub 2021 Dec 22 PubMed.
Goizueta Institute @ Emory Brain Health
The study by Braun et al. is a comprehensive analysis of how different Aβ peptides assemble on their own or when mixed together. These data in general support the notion that though the shorter Aβ peptides can assemble into amyloid fibrils, albeit with slowed rate of kinetics, they delay or inhibit Aβ42 aggregation in the mixtures, and that the complex mixtures of the various peptides inhibit aggregation further.
These data agree with data generated in vivo in preclinical models from my lab and collaborators (Kim et al., 2007; De Mena et al., 2020; Moore et al., 2018) and are also supportive of biomarker studies that suggest that the shorter peptides might be predictive in humans.
It is pretty clear that these types of test tube aggregation studies really do provide good insight, and have a high predictive value for in-vivo effects of Aβ aggregation kinetics.
Where I struggle conceptually is the more general issue that we as a field rarely bring up or discuss openly: Aβ prior to deposition is present at low nanomolar levels and is likely bound to many other proteins, so how does Aβ42 ever nucleate in the brain? Obviously, this takes a long time in humans, but even so we must be missing some aspect of phenomena that occurs in the living brain. Appreciable aggregation ex vivo requires near micromolar concentrations or greater. I personally think there must be some other factor or factors that catalyze the seeding. Alternatively, there must be some way Aβ42 is locally concentrated.
References:
Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007 Jan 17;27(3):627-33. PubMed.
De Mena L, Smith MA, Martin J, Dunton KL, Ceballos-Diaz C, Jansen-West KR, Cruz PE, Dillon KD, Rincon-Limas DE, Golde TE, Moore BD, Levites Y. Aß40 displays amyloidogenic properties in the non-transgenic mouse brain but does not exacerbate Aß42 toxicity in Drosophila. Alzheimers Res Ther. 2020 Oct 17;12(1):132. PubMed.
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
UCL Queen Square Institute of Neurology
University College London
Braun and colleagues present detailed and important kinetic data on Aβ oligomerization in vitro. These are crucial experiments in complex mixes of different Aβ species—Aβ37, Aβ38, Aβ40 and Aβ42—each of which is able to form aggregates and each of which can affect aggregation of other species.
The data distinguishes Aβ42 from the shorter peptides and gives credence to the idea that Aβ42 is a more toxic, aggregation-prone species. Aβ37, Aβ38, and Aβ40 seem to behave similarly to one another, with slower aggregation kinetics. Importantly, while Aβ37, Aβ38, Aβ40 seem to be able to co-aggregate, Aβ42 forms fibrils independently. It will be fascinating to investigate other “toxic” species such as Aβ43 in increasingly more complex Aβ mixes and brain-like environments. For example, it is possible to detect at least 33 different Aβ peptides released from iPSC-derived neurons—could depletion of shorter species promote aggregation (Arber et al., 2019)?
With respect to familial Alzheimer’s disease, it is a crucial finding at Aβ37, Aβ38, and Aβ40 all reduce the aggregation kinetics of Aβ42. Of importance are two aspects: 1) the mixture of Aβ37, Aβ38, and Aβ40 had a greater inhibitor effect on Aβ2 aggregation than each individual peptide and 2) that of the three alloforms, Aβ38 is the more potent inhibitor of Aβ42 aggregation, suggesting that factors other than just length influence inhibition of Aβ42 aggregation. Mutations in PSEN1 are found to reduce γ-secretase processivity of APP/Aβ, increasing the relative production of Aβ42, and reducing the relative production of Aβ37/Aβ38 (Szaruga et al., 2015; Arber et al., 2019; Arber et al., 2019). This paper therefore suggests an uninhibited oligomerization propensity behind Aβ42 aggregation in familial AD.
This then begs the question: What about mutations that increase the entire Aβ spectrum to the same extent? Such as APP duplications or those around the β-secretase cleavage site. Would increasing overall concentration of both Aβ42 and shorter peptides while maintaining the molar ratio promote Aβ42 aggregation?
Finally, it is tempting to speculate on the formation of plaques in nivo. For example, a core could be composed of Aβ42 rich fibrils which then form a surface accelerating aggregation of shorter Aβ species in halos around the core (Saito et al., 2011).
Altogether, this work gives important insights about how Aβ37, Aβ38, and Aβ40 can influence Aβ42 aggregation, and vice versa, and opens a window of opportunities for compounds such as γ-secretase modulators that could favor the production of shorter Aβ alloforms and slow down Aβ42 aggregation.
University of Saskatchewan
γ-Gamma-Secretase-mediated cleavage of amyloid precursor protein (APP) yields a series of length variants, aka alloforms, of the Aβ peptide, including the physiological—and most abundant—Aβ1-40 and the slightly longer Aβ1-42, whose increased production and accumulation within brain and deleterious effects contribute to Alzheimer disease pathology. Shorter alloforms, such as Aβ1-37 and Aβ1-38, also exist in significant relative amounts, and their roles in AD are often presumed to be fibrillogenic and toxic—a naïve extrapolation from the innumerable characterization studies centered on their longer cousins.
This paper by Linse and colleagues relies on the thioflavin T (ThT) binding assay to compare the fibrillogenic behavior of Aβ1-37, Aβ1-38, Aβ1-40, and Aβ1-42 peptides either in isolation or in increasingly complex mixtures. In brief, shorter Aβ alloforms, e.g. Aβ1-37 and Aβ1-38, whether present in the reaction mixture as monomers or as seed fibrils, each can slow the rate of aggregation of either Aβ1-40 or Aβ1-42, and their effect is more pronounced with Aβ1-42 than with Aβ1-40; this confirms some of the observations made previously by us (Quartey et al., 2021) and others (Vandersteen et al., 2012). Linse and colleagues then delve deeper, and the story increases in complexity along with the complexity of the mixtures, with different stages of Aβ1-40 or Aβ1-42 fibril progression being influenced by differing combinations of peptides, and with the effect of Aβ1-38 on Aβ1-42 being more evident than that of either Aβ1-37 or Aβ1-40—something we only hinted at using surface plasmon resonance (Quartey et al., 2021).
These two studies underpin a critical caveat to the amyloid cascade hypothesis of AD; simply put, not all Aβ peptides are bad. This needs to become a mantra for any group that is considering any anti-amyloid therapeutic (immuno-, pharmaco-, etc.) as a clinical management tool; indeed, the current strategy of indiscriminate targeting of Aβ peptides likely explains most of the reported negative AD clinical trial outcomes using anti-amyloids.
A possible solution to this unmet clinical need would be the development of γ-secretase modulators (GSMs). Unlike γ-secretase inhibitors, which reduce the yield of all Aβ peptides to the same extent, GSMs simply shift the cleavage of APP away from Aβ1-42 in favor of Aβ1-37 and Aβ1–38 (e.g. see Czirr et al., 2008; Borgegard et al., 2012; Ahn et al., 2020; Rynearson et al., 2021). Notably, GSMs target APP, while sparing other substrates including Notch. Now that we are aware that these shorter alloforms of Aβ may also provide relief from disease progression, these GSMs are looking even more attractive.
The physiological levels of Aβ1-37 and Aβ1-38 are not inconsequential (~25 percent of total pool of Aβs). Neither is their influence, particularly if one considers (based on the Linse study) that these shorter peptides likely exert most of their effect(s) through reversible—and essentially repeatable—binding. Thus, having any Aβ1-37 and Aβ1-38 will delay Aβ1-42 oligomerization/aggregation sufficiently that Aβ1-42, in equilibrium, would remain predominantly as a small-molecular-weight species. The hope here is that not only would GSMs reduce the effective amount of Aβ1-42, but that any generated, and kept in mono-/dimeric form, would have higher affinity for efflux transport systems/clearance from the brain and/or be in an optimal conformation for enzymatic degradation.
Parenthetically, it will be interesting to determine whether Aβ1-37 and Aβ1-38 are candidates for the same efflux systems and/or degradative pathways as their longer cousins, or whether they can allosterically modulate recognition of the longer species by transport and/or enzymatic systems, thus potentiating benefit for the AD brain.
A recent paper found that the higher the concentration of Aβ1-38 in CSF, the lower the likelihood of a diagnosis of dementia on follow-up (four years) (Cullen et al., 2021). Our own work, based on a limited sample size of autopsied brain samples, revealed that a higher ratio of soluble Aβ1-42/Aβ1-38 correlated with earlier age-at-death in older donors with a confirmed diagnosis of AD; this held true for males, but not females (Quartey et al., 2021). An earlier study of ours using the same autopsy samples found a negative correlation between nicastrin (a component of the γ-secretase complex) and insoluble Aβ1-42 in males, but a positive correlation in females (Nyarko et al., 2018). Similar sex-specific observations had previously been made in brain of a mouse model of AD (Placanica et al., 2009). Thus, any GSM-based management options in the clinic will have to consider the very strong likelihood of sex-dependent benefit.
Clearly, the generation of a pool of Aβ peptide alloforms is not simply a generalized maladaptive response in a diseased brain. The brain is said to be able to fix itself; in the case of AD, it appears to have chosen to pit Aβ peptide against Aβ peptide, and GSMs that target Aβ1-42 while promoting Aβ1-37 and Aβ1-38 may provide the leverage needed in delaying onset of disease in those most at risk.
References:
Quartey MO, Nyarko JN, Maley JM, Barnes JR, Bolanos MA, Heistad RM, Knudsen KJ, Pennington PR, Buttigieg J, De Carvalho CE, Leary SC, Parsons MP, Mousseau DD. The Aβ(1-38) peptide is a negative regulator of the Aβ(1-42) peptide implicated in Alzheimer disease progression. Sci Rep. 2021 Jan 11;11(1):431. PubMed.
Vandersteen A, Masman MF, De Baets G, Jonckheere W, Van Der Werf K, Marrink SJ, Rozenski J, Benilova I, De Strooper B, Subramaniam V, Schymkowitz J, Rousseau F, Broersen K. Molecular Plasticity Regulates Oligomerization and Cytotoxicity of the Multipeptide-length Amyloid-β Peptide Pool. J Biol Chem. 2012 Oct 26;287(44):36732-43. PubMed.
Czirr E, Cottrell BA, Leuchtenberger S, Kukar T, Ladd TB, Esselmann H, Paul S, Schubenel R, Torpey JW, Pietrzik CU, Golde TE, Wiltfang J, Baumann K, Koo EH, Weggen S. Independent generation of Abeta42 and Abeta38 peptide species by gamma-secretase. J Biol Chem. 2008 Jun 20;283(25):17049-54. PubMed.
Borgegard T, Juréus A, Olsson F, Rosqvist S, Sabirsh A, Rotticci D, Paulsen K, Klintenberg R, Yan H, Waldman M, Stromberg K, Nord J, Johansson J, Regner A, Parpal S, Malinowsky D, Radesater AC, Li T, Singh R, Eriksson H, Lundkvist J. First and Second Generation γ-Secretase Modulators (GSMs) Modulate Amyloid-β (Aβ) Peptide Production through Different Mechanisms. J Biol Chem. 2012 Apr 6;287(15):11810-9. PubMed.
Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, Mendes da Costa L, Qiu R. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin Pharmacol Ther. 2020 Jan;107(1):211-220. Epub 2019 Sep 11 PubMed.
Rynearson KD, Ponnusamy M, Prikhodko O, Xie Y, Zhang C, Nguyen P, Hug B, Sawa M, Becker A, Spencer B, Florio J, Mante M, Salehi B, Arias C, Galasko D, Head BP, Johnson G, Lin JH, Duddy SK, Rissman RA, Mobley WC, Thinakaran G, Tanzi RE, Wagner SL. Preclinical validation of a potent γ-secretase modulator for Alzheimer's disease prevention. J Exp Med. 2021 Apr 5;218(4) PubMed.
Cullen N, Janelidze S, Palmqvist S, Stomrud E, Mattsson-Carlgren N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative. Association of CSF Aβ38 Levels With Risk of Alzheimer Disease-Related Decline. Neurology. 2022 Mar 1;98(9):e958-e967. Epub 2021 Dec 22 PubMed.
Nyarko JN, Quartey MO, Pennington PR, Heistad RM, Dea D, Poirier J, Baker GB, Mousseau DD. Profiles of β-Amyloid Peptides and Key Secretases in Brain Autopsy Samples Differ with Sex and APOE ε4 Status: Impact for Risk and Progression of Alzheimer Disease. Neuroscience. 2018 Mar 1;373:20-36. Epub 2018 Jan 11 PubMed.
Placanica L, Zhu L, Li YM. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One. 2009;4(4):e5088. PubMed.
Make a Comment
To make a comment you must login or register.