Fueled by Fat: Lipid Droplets a Critical Energy Source for Synapses
Quick Links
To satisfy the incessant energy demands of their thousands of synapses, neurons have to have enormous supplies of ATP at the ready. Where does this seemingly bottomless well of energy come from? It’s lipid droplets stationed within pre-synaptic terminals, according to a study published July 1 in Nature Metabolism. Scientists led by Timothy Ryan of Weill Cornell Medical College in New York report that synaptic activity spurs the breakdown of triglycerides stored within the droplets into fatty acids, which synaptic mitochondria promptly convert into ATP. DDHD2—a neuronal enzyme that converts triglycerides into fatty acids—facilitates this process. Without it, a glut of lipid droplets soon clogged pre-synapses, snuffing firing to a flicker. Pinching off this synaptic fat supply was enough to put mice into a torpor.
- The triglyceride lipase DDHD2 catabolizes lipids stored in droplets within pre-synapses.
- When neurons fire, mitochondria use the fatty acids to generate ATP.
- Without this fat, synaptic function falters, putting mice into torpor.
- The findings challenge the idea that synaptic firing is fueled primarily by glucose.
The findings challenge the long-held notion that neurons, presumably to conserve precious lipids for their copious membranes, prefer to use glucose for energy instead of fat, commented Russell Swerdlow of Kansas University Medical Center. “This paper turns that idea on its head, or at least indicates that the broad assumption may go too far,” he wrote. “Here, it certainly seems that some neuron populations can, and perhaps eagerly do, pursue β-oxidation.” This biochemical process converts fatty acids into energy in the form of ATP by removing two-carbon acetyl groups sequentially from the lipid chain, hence the “β”; the acetyl groups then enter the citric acid cycle. This takes place in mitochondria.
“The idea that fatty acid oxidation occurs at synapses to meet their energetic demands certainly challenges previous studies, but it makes sense,” wrote Amita Sehgal of the University of Pennsylvania in Philadelphia. “While most work on brain lipids has focused on glia, going forward we will have to take this neuronal role into account,” she added.
Indeed, multiple recent studies, including Sehgal’s, have spotted lipid droplets in microglia and astrocytes, where the fatty blobs are thought to sequester excess fat produced by stressed neurons, for example in models of amyloidosis and tauopathy (Feb 2024 news; Mar 2024 news; Apr 2024 news). In other tissues, lipid droplets reportedly serve as storage depots for triglycerides, which can be converted to fatty acids and then transformed into ATP via β-oxidation in mitochondria.
Lipid droplets have rarely been spotted within neurons, reinforcing the notion that neurons don’t metabolize fat for energy. However, in a rare disorder called hereditary spastic paraplegia, which is caused by a mutation in a neuronal triglyceride lipase DDHD2, the droplets do clog neurons (Schuurs-Hoeijmakers et al., 2012; Inloes et al., 2014). This hinted to the researchers that, under healthy conditions, neurons might burn through these reserves before they can even be seen.
To investigate the role of these lipid droplets in neuronal metabolism, first author Mukesh Kumar and colleagues started with a closer look at DDHD2. Via immunostaining, they spotted the lipase in neurons throughout the mouse brain, where it was concentrated in axonal terminals. Treating mice with the selective DDHD2 inhibitor KLH45 for four days ramped up triglycerides in the brain. In cultured hippocampal neurons, DDHD2 inhibition instigated a fivefold increase in neuronal lipid droplets, which accumulated in neuronal pre-synapses alongside resident mitochondria. The same happened when the researchers supplemented the neurons with excess fatty acids, suggesting that neurons catabolize lipids in these synaptic droplets only in times of need.
Droplets also promptly accumulated when the researchers shut down synaptic firing with the sodium channel blocker tetrodotoxin, suggesting that the lipid droplets fuel synaptic transmission (image below). This synaptic shushing also shut down the catabolism of triglycerides into fatty acids, their transfer to mitochondria, and the production of ATP via β-oxidation.
Is this “biodiesel” a supplement or an alternative to glucose? Even in the complete absence of the latter, this fatty fuel maintained synaptic ATP at near-normal levels. This was sufficient to support the most energy-intensive synaptic functions, such as rapid recycling of synaptic vesicles in response to repeated action potentials.
In sum? “These findings demonstrate that axonal mitochondria can metabolize fatty acids to sustain ATP production during glucose scarcity and that this metabolic pathway is dynamically regulated by neuronal activity,” concluded Sunan Li and Zu-Hang Sheng of the National Institutes of Health in a Nature News & Views.
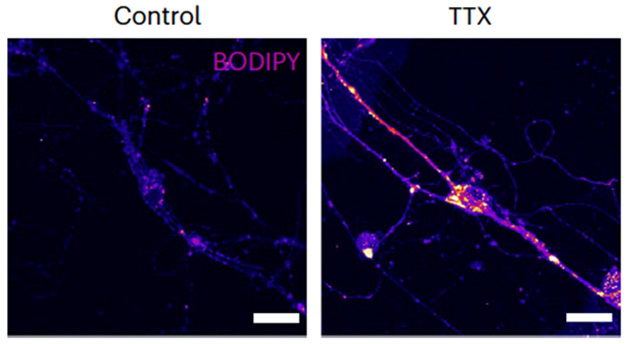
Lipid Gridlock. Neurons have few lipid deposits (left), but blocking neuronal activity with tetrodotoxin (right) results in a buildup of lipids within synaptic terminals as well as in the soma (yellow, magenta). [Courtesy of Kumar et al., Nature Metabolism, 2025.]
Finally, the scientists hypothesized that shutting down this metabolic pathway in mice might trigger torpor—a hypothermic state induced by a combination of metabolic stresses such as starvation and cold. Torpor is regulated by neurons in the hypothalamus. The scientists first deprived mice of food for three hours, then injected them with a DDHD2 inhibitor. Three hours later, the animals’ body temperatures had plunged by nearly 7oC, while mice who were merely deprived of food were only 1oC colder.

Fat on Demand. Spurred by action potentials, DDHD2 within the pre-synapse catabolizes lipid droplets into fatty acids, which mitochondria then convert via β-oxidation to ATP (left). Without active DDHD2, lipid droplets swell and fatty acid metabolism stalls, derailing synaptic function and putting mice into torpor (right). [Courtesy of Kumar et al., Nature Metabolism, 2025.]
Where do the lipids that go into the droplets come from? And what molecules drive this supply chain? The authors speculated that ApoE could deliver these lipids, packaged into lipoprotein particles, to neurons, which then use lipoprotein lipase to extract triglycerides from the particles for storage in droplets. The potential role of ApoE in this pathway suggests it could be relevant to synaptic function in AD. Li and Sheng noted that ApoE4, in particular, hampers neuronal uptake of long-chain fatty acids by disrupting recycling of the lipoprotein receptor SORT1 (Asaro et al., 2021; Greta et al., 2024). “Whether such ApoE4-related defects in neuronal fatty acid metabolism contribute to synaptic dysfunction and cognitive decline remains to be explored,” they added.
Priyanka Narayan of the National Institutes of Health in Bethesda, Maryland, believes the findings shift our understanding of how neurons meet their energetic demands. She wondered how this might change with age. “Does neuronal reliance on β-oxidation become more pronounced in conditions of limited glucose, such as during aging or in disease states?” she asked. In such cases, problems with fatty acid metabolism might promote neurodegeneration, suggested Ryan. Narayan thinks the findings invite broader investigation of fatty acid metabolism in neurons (comment below).—Jessica Shugart
References
News Citations
- While a Fly Sleeps, Its Glia Burn Neuronal Lipids to Refresh the Brain
- Paper Alert: APOE4 Packs on Lipid Droplets in Microglia
- Stirred by Tau, Neurons Amp Up Lipid Droplets in Glia
Paper Citations
- Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B, van de Vondervoort II, van der Graaf M, Nobau AC, Otte-Höller I, Vermeer S, Smith AC, Humphreys P, Schwartzentruber J, FORGE Canada Consortium, Ali BR, Al-Yahyaee SA, Tariq S, Pramathan T, Bayoumi R, Kremer HP, van de Warrenburg BP, van den Akker WM, Gilissen C, Veltman JA, Janssen IM, Vulto-van Silfhout AT, van der Velde-Visser S, Lefeber DJ, Diekstra A, Erasmus CE, Willemsen MA, Vissers LE, Lammens M, van Bokhoven H, Brunner HG, Wevers RA, Schenck A, Al-Gazali L, de Vries BB, de Brouwer AP. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012 Dec 7;91(6):1073-81. Epub 2012 Nov 21 PubMed.
- Inloes JM, Hsu KL, Dix MM, Viader A, Masuda K, Takei T, Wood MR, Cravatt BF. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A. 2014 Oct 14;111(41):14924-9. Epub 2014 Sep 29 PubMed.
- Asaro A, Sinha R, Bakun M, Kalnytska O, Carlo-Spiewok AS, Rubel T, Rozeboom A, Dadlez M, Kaminska B, Aronica E, Malik AR, Willnow TE. ApoE4 disrupts interaction of sortilin with fatty acid-binding protein 7 essential to promote lipid signaling. J Cell Sci. 2021 Oct 15;134(20) Epub 2021 Oct 21 PubMed.
- Willnow T, Greda AK, Gomes JP, Zurawska-Plaksej E, RaphaelaFritsche-Guenther R, Rudolph I-M, Telugu NS, Coemert C, Kirwan J, Kunz S, Rothe M, Diecke S, Bross P. Interaction of sortilin with apolipoprotein E3 enables neurons to use long-chain fatty acids as alternative metabolic fuel. 2024 Jun 10 10.1101/2024.06.10.598173 (version 1) bioRxiv.
Further Reading
No Available Further Reading
Primary Papers
- Kumar M, Wu Y, Knapp J, Pontius CL, Park D, Witte RE, McAllister R, Gupta K, Rajagopalan KN, De Camilli P, Ryan TA. Triglycerides are an important fuel reserve for synapse function in the brain. Nat Metab. 2025 Jul 1; Epub 2025 Jul 1 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Kansas
This is a very interesting study that challenges some existing conceptions and brings to the fore questions about brain energy metabolism. It counters a pre-existing notion that neurons are not enthusiastic about catabolizing fatty acids through mitochondrial -oxidation to generate energy, presumably because neurons require a lot of membrane production and it may behoove neurons not to catabolize fatty acids. This paper turns that idea on its head, or at least indicates that broad assumptions may go too far.
Here, it certainly seems that some neuron populations, in some instances (for example at active synapses) can, and perhaps eagerly do, pursue β-oxidation. One might argue that these experiments are in some ways susceptible to the usual limitations that accompany the use of model systems, and some data streams used to argue the conclusions could have benefitted from added rigor. One example would be confirming that the neutral lipid deposits referred to as lipid droplets are in fact lipid droplets, as opposed to lysosomal lipid.
Technical caveats aside, and although this was not specifically designed as an “AD study,” it has AD ramifications. It provides some insight into lipid accumulations in AD brains. It emphasizes a point that I think is quite central to AD, which is that it is virtually impossible to disentangle lipid and mitochondrial metabolism from each other. It raises a very interesting point about a cycle called the “Randle Cycle,” which I had not previously considered. The Randle cycle refers to a reciprocal relationship between glucose and lipid-fueled bioenergetic activity. It is interesting to consider what such a balance looks like in the AD brain.
Finally, this paper pulls into the discussion ApoE, and presents what I think is a reasonable angle from which to consider ApoE’s mechanistic connection to AD.
Overall, this is a very interesting read.
National Institutes of Health
This study provides compelling evidence that neurons can use triglyceride-rich lipid droplets to support synaptic activity. This work challenges the long-held view that neurons rely exclusively on glucose, and it reveals a previously underappreciated flexibility in neuronal bioenergetics.
Using a combination of elegant neuronal stimulation and inhibitor experiments, the authors show that activity-induced breakdown of synaptic lipid droplets via the lipase DDHD2 fuels mitochondrial β-oxidation. Extending these findings in vivo, this study demonstrates that neuronal β-oxidation is essential to prevent a torpor-like state in mice.
These findings shift our understanding of how neurons meet their energetic demands and open important mechanistic and physiological questions. What molecular pathways link neuronal activity to lipid droplet lipolysis and fatty acid import into mitochondria? Does neuronal reliance on β-oxidation become more pronounced in conditions of limited glucose, such as during aging or in disease states? And do alterations in lipid droplet dynamics—either within neurons or in neighboring glia, potentially involving intercellular lipid transfer—modulate this metabolic flexibility?
With lipid droplets gaining increasing attention in the context of multiple neurodegenerative diseases, these findings invite broader consideration of how fatty acid metabolism may shape neuronal function and vulnerability, beyond the context of hereditary spastic paraplegias.
Brigham & Women's Hospital/Harvard Medical School
Healthy neurons provide a stubborn environment for lipid droplets (LDs) to thrive, due to the cellular machinery that enables rapid synthesis and degradation of triglycerides, a principal component of LDs. Decreasing the activity of the neuronal lipase DDHD2, which is responsible for the accelerated catabolism of triglycerides, results in the accumulation of LDs in otherwise healthy neurons. We have adapted this approach to investigate whether triglyceride-rich LDs influence the physiological states of α-synuclein (Bolsinger et al., 2025), an abundant brain protein involved in regulating neurotransmission.
Though neurons heavily depend on glucose for energy, under hypoglycemic states they utilize ketones, a precursor for lipid synthesis in the brain, to meet their energy demands. Here, the Ryan lab combined KLH45, a small-molecule inhibitor of DDHD2, with multiple approaches to elegantly demonstrate that neuronal lipids, particularly triglyceride-rich LDs, can efficiently serve as a fuel source for synaptic vesicle recycling during hypoglycemia.
Some open questions that remain include:
References:
Bolsinger MM, Moors TE, Brontesi L, Nuber S, Dettmer U, Ramalingam N. Acute lipid droplet accumulation induced by the inhibition of the phospholipase DDHD2 does not affect the level, solubility, or phosphoserine-129 status of α-synuclein. Metab Brain Dis. 2025 Jan 24;40(1):111. PubMed.
Henan Academy of Innovations in Medical Science
These findings are very interesting in the context of Alzheimer’s disease. We know that glucose utilization is impaired in AD brains. 18FDG-PET scans have demonstrated impaired glucose uptake in individuals with mild cognitive impairment (MCI), early AD, and a family history of developing AD (Edison et al., 2008; Mosconi et al., 2010; Bailly et al., 2015). In fact, these hypometabolic changes precede the appearance of dementia symptoms by a decade or more (Raut et al., 2023; Yonamine et al., 2023; de la Monte, 2024).
In order to compensate for loss of glucose utilization, lipids are broken down to ketones by astrocytes and "shipped" to neurons to be directly turned into ATP by mitochondria without the need for glycolysis. If the supply of ketones is inadequate, glia cells can break down the myelin sheath of axons in white-matter sections of the brain, which is made of lipids, to produce ketones. This is the reason why white-matter loss is found in AD brains (Makino et al., 2014; Yassine et al., 2022). The lipid stores in glia can be seen under the microscope as lipid droplets. In fact, the earliest evidence of disturbed energy utilization in the brain came from Alois Alzheimer’s description of the first AD brain tissue analysis, where he noticed that cells contained lipid droplets that are not normally seen in the brain (Alzheimer et al., 1995; Foley, 2010).
APOE is a lipid transporter in the brain and we know that the allele ApoE ε4 is a risk factor for AD (Corder et al., 1993). Studies showed that the levels of cholesterol and LDL lipid transporters is changed in carriers of the ApoE ε4 allele and even more so in those who have developed AD (Xu et al., 2023). Importantly, LDL lipid transporters in the CSF are smaller and contain lower levels of lipids in ApoE ε4 carriers (Safieh et al., 2019), and brain tissue analysis of ApoE ε4 carriers show clear impairments in lipid metabolism (Nock et al., 2017).
Therefore, the reason why ApoE ε4 carriers are at risk to develop AD is most likely due to the fact that ApoE ε4 is less able to transport lipids in the brain to compensate for the loss of glucose utilization in neurons. If synapses use lipids/ketones for energy, reduced supplies of ketones by ApoE ε4 will most certainly accelerate synaptic failure and disease progression.
References:
Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde". Clin Anat. 1995;8(6):429-31. PubMed.
Bailly M, Destrieux C, Hommet C, Mondon K, Cottier JP, Beaufils E, Vierron E, Vercouillie J, Ibazizene M, Voisin T, Payoux P, Barré L, Camus V, Guilloteau D, Ribeiro MJ. Precuneus and Cingulate Cortex Atrophy and Hypometabolism in Patients with Alzheimer's Disease and Mild Cognitive Impairment: MRI and (18)F-FDG PET Quantitative Analysis Using FreeSurfer. Biomed Res Int. 2015;2015:583931. Epub 2015 Jun 17 PubMed.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993 Aug 13;261(5123):921-3. PubMed.
de la Monte SM. Conquering Insulin Network Dysfunctions in Alzheimer's Disease: Where Are We Today?. J Alzheimers Dis. 2024;101(s1):S317-S343. PubMed.
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008 Dec;32(3):412-9. PubMed.
Foley P. Lipids in Alzheimer's disease: A century-old story. Biochim Biophys Acta. 2010 Aug;1801(8):750-3. PubMed.
Makino T, Umegaki H, Suzuki Y, Yanagawa M, Nonogaki Z, Nakashima H, Kuzuya M. Relationship between small cerebral white matter lesions and cognitive function in patients with Alzheimer's disease and amnestic mild cognitive impairment. Geriatr Gerontol Int. 2013 Nov 12; PubMed.
Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis. 2010;20(3):843-54. PubMed.
Nock TG, Chouinard-Watkins R, Plourde M. Carriers of an apolipoprotein E epsilon 4 allele are more vulnerable to a dietary deficiency in omega-3 fatty acids and cognitive decline. Biochim Biophys Acta. 2017 Oct;1862(10 Pt A):1068-1078. Epub 2017 Jul 18 PubMed.
Raut S, Bhalerao A, Powers M, Gonzalez M, Mancuso S, Cucullo L. Hypometabolism, Alzheimer's Disease, and Possible Therapeutic Targets: An Overview. Cells. 2023 Aug 8;12(16) PubMed.
Safieh M, Korczyn AD, Michaelson DM. ApoE4: an emerging therapeutic target for Alzheimer's disease. BMC Med. 2019 Mar 20;17(1):64. PubMed.
Xu H, Fu J, Mohammed Nazar RB, Yang J, Chen S, Huang Y, Bao T, Chen X. Investigation of the Relationship between Apolipoprotein E Alleles and Serum Lipids in Alzheimer's Disease: A Meta-Analysis. Brain Sci. 2023 Nov 6;13(11) PubMed.
Yassine HN, Solomon V, Thakral A, Sheikh-Bahaei N, Chui HC, Braskie MN, Schneider LS, Talbot K. Brain energy failure in dementia syndromes: Opportunities and challenges for glucagon-like peptide-1 receptor agonists. Alzheimers Dement. 2022 Mar;18(3):478-497. Epub 2021 Oct 14 PubMed.
Yonamine CY, Michalani ML, Moreira RJ, Machado UF. Glucose Transport and Utilization in the Hippocampus: From Neurophysiology to Diabetes-Related Development of Dementia. Int J Mol Sci. 2023 Nov 18;24(22) PubMed.
Make a Comment
To make a comment you must login or register.