Inside Out—Plaques May Have Intracellular Origin
Quick Links
Since amyloid-β plaques are extracellular and neurons churn out the plaques’ constituent peptides, researchers have, by and large, assumed that Aβ starts aggregating outside the cell. However, two recent studies bolster the idea that events kicking off plaque formation may, in fact, occur within cells. Applying state-of-the-art biophysics to tackle the cell biology of Aβ aggregation, German scientists have developed a cell culture system for modeling Aβ plaque formation. Reported online in this week’s PNAS Early Edition, their data suggest that cells internalize Aβ peptides and shunt them to intracellular vesicles, where Aβ gathers into fibrils that can rupture vesicular membranes. In this model, these events eventually choke the life out of cells, causing them to spew their clumps of intracellular amyloid into the extracellular space. “Our mechanism reconciles the extracellular starting point (i.e., Aβ peptides) and endpoint (i.e., extracellular plaques) with an intermediate step that occurs inside the cell,” lead investigator Marcus Fändrich of Max-Planck Research Unit for Enzymology of Protein Folding in Halle, Germany, told ARF.
A similar story emerged from a study published in the December 1, 2009, PNAS. In that paper, Jin-Moo Lee and colleagues at Washington University School of Medicine, St. Louis, Missouri, show that cells can take up Aβ peptides at low extracellular concentrations and concentrate them in late endosomes/lysosomes, where they form aggregates capable of seeding growth of amyloid fibrils. By highlighting the potential importance of intracellular events in the initial steps of plaque formation, the two studies may help guide the development of AD drugs that target Aβ aggregation.
The claim that plaque formation may have an intracellular basis is not new. Analyses of brain tissue from AD patients and various AD mouse models have found Aβ in multivesicular bodies (MVBs) (Takahashi et al., 2002; Oakley et al., 2006; and ARF related news story) and in endosomes (Rajendran et al., 2007). Furthermore, amyloid plaques routinely contain lysosomal proteases and other proteins originally found inside the cell (Cataldo et al., 1990; Wilhelmus et al., 2007). However, debate continues over whether intracellular or extracellular Aβ primarily causes AD, and how.
To gain some insight into these issues, Fändrich’s team set out to devise a method that would enable careful scrutiny of plaque formation in a culture dish. The current study builds upon a system his lab had previously developed using primary human macrophages to make Aβ plaques (Gellermann et al., 2006). “We have substantially improved it and made it more reliable,” Fändrich said.
First author Ralf Friedrich and colleagues sprinkled Aβ(1-40) peptide into the culture medium of a handful of different cell types, and found that the two monocytic lines—one mouse (J-774A.1), one human (THP-1)—produced the greatest plaque load. Human neuroblastoma cells (SHSY5Y), and mouse and human primary phagocytes could also do the job, though not nearly as robustly. The researchers used THP-1 cells for most of the remaining experiments. They confirmed they were seeing bona fide Aβ plaques using a host of biochemical, biophysical, and immunostaining techniques.
When they threw in paraformaldehyde or fixed with methanol to kill the cells, plaques did not form, suggesting that living cells are required for the process. This came as a surprise. “You can take pure Aβ peptide and put it in a test tube, and it will form amyloid fibers. So you would not expect that cells need to be present to form plaques,” Fändrich said. “Yet we reproducibly found that when it comes to formation of fibrils and plaques, cells are indeed involved.” Time-lapse video microscopy using fluorescent Aβ(1-40) confirmed this, showing plaques always forming right next to cells or within cell bodies.
Addition of endocytosis and phagocytosis inhibitors (cytochalasin B or latrunculin B) also reduced plaque yield, suggesting that Aβ peptides may need to be internalized before they form plaques. However, Aβ taken up by THP-1 cells did not co-localize with early endosomal or lysosomal markers, as revealed by freeze-fracturing the cells and peering at their insides with Aβ immunogold labeling and transmission electron microscopy. Instead, the researchers saw Aβ forming fibrillar bundles within MVBs; these are multifunctional compartments that interface with other vesicular pathways, including the lysosome/endosomal system. Even more stunning, some of the Aβ fibrils looked as though they had punctured the MVB membrane (see image). “Initially we thought this could be an artifact,” Fändrich said. “But we observed it with many cells. It’s something we saw reproducibly.”
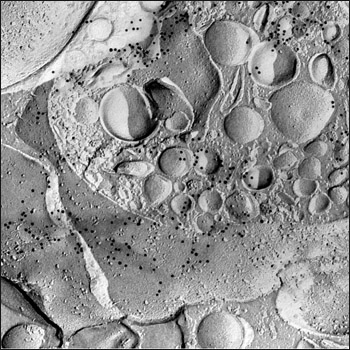
Piercing Through Vesicles
Transmission electron microscopy shows Aβ peptides (small black dots) lining up to form fibrillar bundles (dark grey shadow) protruding from a multivesicular body (upper right corner). Image credit: Martin Westermann, Ralf P. Friedrich, Marcus Fändrich
One caveat is that the model may not directly capture what happens in the brain and in neurons. “But it’s a step forward,” said Gunnar Gouras of Weill Cornell Medical College in New York. “We can study β amyloid in a test tube, and we can do pathology in the brain, and it's not easy to combine those two, but that's what they're doing.” (See full comment below.)
Fändrich said their working hypothesis is that internalization of Aβ peptides into intracellular vesicles promotes their aggregation not only by increasing local Aβ concentration but also by presenting factors (e.g., gangliosides) known to encourage fibrillation.
The Washington University scientists arrived at a similar mechanism “but with different models that may be telling different parts of the same story,” Lee told ARF. His team designed a study to address a longstanding conundrum about Aβ—namely, that Aβ42, the major form in AD plaques, assembles into fibrils in vitro at high (micromolar) concentrations and in acidic conditions not found in the brain’s extracellular fluid. Such circumstances do exist within late endosomes/lysosomes.
To visualize Aβ internalization, first author Xiaoyan Hu and colleagues incubated fluorescent Aβ42 with SHSY5Y human neuroblastoma cells. Confocal microscopy revealed cells taking up Aβ within 24 hours at physiologically relevant concentrations of extracellular Aβ as low as 1 nM. Furthermore, the researchers found Aβ-containing vesicles co-staining with lysosomal markers, suggesting that Aβ gets trafficked into the lysosomal pathway. It appears to get cleared out quickly, though, as only a few fluorescent vesicles remained 24 to 48 hours after washout with Aβ-free medium.
Performing pixel-by-pixel analysis of fluorescence intensity to analyze 29 vesicles from five different images, the researchers estimated that vesicular Aβ was concentrated about 100-fold relative to its extracellular starting conditions. Furthermore, the intracellular Aβ formed aggregates that were able to seed formation of amyloid fibrils when mixed for 48 hours with fluorescent Aβ(1-42) at concentrations normally too low for spontaneous fibril formation.
A key difference between the two studies concerns which vesicular compartment Aβ peptides went to once the cell had taken them in from the outside. “I think these are differences in terms of how the Aβ is taken up and how it’s trafficked,” Lee said, noting these could have stemmed from the different cell types (monocyte vs. neuronal) analyzed by each group. “In my case, we’re seeing receptor-mediated endocytosis, which goes to a lysosome. I think [Fändrich] may be seeing more phagocytosis, which is not receptor-mediated.” Fändrich’s observations seemed reminiscent of several recent papers demonstrating phagocytosis of tau and polyglutamine fibrils (Frost et al., 2009; Ren et al., 2009), Lee said.—Esther Landhuis
References
News Citations
Paper Citations
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006 Oct 4;26(40):10129-40. PubMed.
- Rajendran L, Knobloch M, Geiger KD, Dienel S, Nitsch R, Simons K, Konietzko U. Increased Abeta production leads to intracellular accumulation of Abeta in flotillin-1-positive endosomes. Neurodegener Dis. 2007;4(2-3):164-70. PubMed.
- Cataldo AM, Thayer CY, Bird ED, Wheelock TR, Nixon RA. Lysosomal proteinase antigens are prominently localized within senile plaques of Alzheimer's disease: evidence for a neuronal origin. Brain Res. 1990 Apr 16;513(2):181-92. PubMed.
- Wilhelmus MM, de Waal RM, Verbeek MM. Heat shock proteins and amateur chaperones in amyloid-Beta accumulation and clearance in Alzheimer's disease. Mol Neurobiol. 2007 Jun;35(3):203-16. PubMed.
- Gellermann GP, Ullrich K, Tannert A, Unger C, Habicht G, Sauter SR, Hortschansky P, Horn U, Möllmann U, Decker M, Lehmann J, Fändrich M. Alzheimer-like plaque formation by human macrophages is reduced by fibrillation inhibitors and lovastatin. J Mol Biol. 2006 Jul 7;360(2):251-7. PubMed.
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009 May 8;284(19):12845-52. PubMed.
- Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009 Feb;11(2):219-25. PubMed.
Further Reading
Papers
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. PubMed.
Primary Papers
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20324-9. PubMed.
- Friedrich RP, Tepper K, Rönicke R, Soom M, Westermann M, Reymann K, Kaether C, Fändrich M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A. 2010 Feb 2;107(5):1942-7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UTMB
This is an interesting study. The ability to visualize intracellular accumulation of Aβ after 24-hour incubation with very low extracellular Aβ (1 nm) is remarkable. The internalization seems to be sequence-specific; this would require a receptor (still unknown) with high affinity to Aβ.
The study also demonstrates that internalized Aβ formed HMW aggregates. Moreover, these aggregates are capable of seeding Aβ fibril formation in vitro. The fate of these aggregates remains uncertain based on this paper. Do they get released back into the media by exocytosis?
View all comments by Rakez KayedLund University
This is a very intriguing study by Fändrich, Friedrich, and colleagues modeling plaque formation in cultured cells following addition of exogenous Aβ. The methods used, including their EM, are excellent. Work such as this is important, while the continued reluctance to even consider the intracellular aspect of Aβ pathology is holding back the field. Leakage of Aβ from multivesicular bodies (MVBs) certainly is also consistent with what we observed by EM in brain. At the same time, the long trails of this Aβ are surprising. Cell culture model systems like the ones used in this paper have many advantages in studying the biological mechanisms of disease.
I would add a few points to their discussion. Light and electron microscopy evidence supports that Aβ42 accumulation begins in neurons and particularly their distal neurites and synapses. The latter can help explain why one can have many plaques with little cell death. At the same time, our studies, as well as very nice work by D’Andrea and colleagues in human brain, and work by Vassar, Bayer, and others in AD transgenic mice, support that plaques also develop from neuron cell bodies. The current work additionally highlights the potential importance of glia in plaque formation. This study emphasizes Aβ internalization in plaque formation, although it is possible that not much Aβ internalization may be needed to upregulate an intracellular pool of Aβ that already is present in MVBs.
View all comments by Gunnar GourasUniversity of Pennsylvania
These are very interesting studies of a topic that merits further investigation.
It is noteworthy that earlier mRNA expression profile data support this notion. Briefly, Steve Ginsberg and colleagues (Ginsberg et al., 1999) followed up on two of our prior studies showing that RNA is sequestered in AD senile plaques (Ginsberg et al., 1997) and that Aβ is detected inside neurons (Wertkin et al., 1993). Thus, Ginsberg et al. analyzed the mRNA profile in single immunocytochemically identified plaques in sections of AD hippocampus. By using amplified RNA expression profiling, polymerase chain reaction, and in situ hybridization, Ginsberg et al. assessed the presence and abundance of 51 mRNAs that encode proteins implicated in the pathogenesis of AD. He compared the mRNAs in amyloid plaques with those in individual CA1 neurons and the surrounding neuropil of control subjects. Remarkably, Ginsberg et al. demonstrated that neuronal mRNAs predominated in amyloid plaques, thereby suggesting that these mRNAs are components of plaques and that these mRNAs may interact with Aβ released from dying neurons when plaques form at sites where neurons degenerate in the AD brain.
References:
Ginsberg SD, Crino PB, Hemby SE, Weingarten JA, Lee VM, Eberwine JH, Trojanowski JQ. Predominance of neuronal mRNAs in individual Alzheimer's disease senile plaques. Ann Neurol. 1999 Feb;45(2):174-81. PubMed.
Ginsberg SD, Crino PB, Lee VM, Eberwine JH, Trojanowski JQ. Sequestration of RNA in Alzheimer's disease neurofibrillary tangles and senile plaques. Ann Neurol. 1997 Feb;41(2):200-9. PubMed.
Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, Lee VM. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A. 1993 Oct 15;90(20):9513-7. PubMed.
Yale University School of Medicine
Two things about this study puzzle me. I don’t understand why the uptake of Aβ peptides by macrophages in culture necessarily mimics the development of plaques in the brains of AD patients, or why it is even a good model system.
The claim that internalized amyloid fibrils penetrate multivesicular membranes is a provocative one, but I question whether the immunostained, freeze-cleaved images that the authors provide are strong enough evidence to support this claim. To obtain these images, platinum-carbon replicas of cleaved cells were “washed” in SDS and then exposed sequentially to anti-Aβ antibodies and gold-labeled anti IGG. The assumption is made that the Aβ antigens in the cell adhere to the replicas, and remain in place during the washes and incubations. How much of the original antigenic material remains adherent to the replicas? Could some of it redistribute during the processing of the replica? The images of the vesicle membranes do not show obvious breaks, but these may be hard to recognize in these preparations. If such large clumps of amyloid fibrils do indeed stream out of multivesicular bodies, they should be easy to demonstrate by conventional thin-sectioning EM.
Rutgers - New Jersey Medical School
Two papers published recently in PNAS (1,2) touch an issue close to our hearts: the nucleation of neuritic plaques in Alzheimer disease. The most important issue about the seeded polymerization hypothesis (3) at this time is the nature of the “bad” seed that could nucleate polymerization of soluble Aβ below the critical concentration. Where does the seed originate? How is it produced? Several years ago, we proposed that these seeds are generated inside specific populations of neurons, and that they accumulate at the neurite terminals. We described a culture system where CNS-derived neuronal cells (CAD) accumulate within their neurites oligomeric Aβ (4). We also proposed that these Aβ aggregates somehow become extracellular (e.g., by cell death, or through fusion of Aβ-containing compartments with the plasma membrane), and provided evidence that the aggregates are indeed externalized (5). In this way, they could readily nucleate the polymerization of the soluble Aβ present in the extracellular space. The question was where the Aβ that we detected inside the neurites comes from.
Our data suggested that the intracellular Aβ aggregates reside in the late endosomes or autophagic vacuoles, since the oligomeric Aβ was detected in large vesicular structures that stain positive for Rab7, a marker for late endosomes and autophagic vacuoles. We also asked whether the neuritic Aβ aggregates could originate from the Aβ present in the culture medium that is taken up by cells via endocytosis. While we clearly showed that reuptake of Aβ via endocytosis does occur, the internalized Aβ accumulates in the cell body, not within the neurites (4), a result that is similar to what the featured papers now report (1,2).
We concluded that the Aβ aggregates present in the cell body and those that accumulate in the neurites likely have a different origin. We still think that most plaques are nucleated from neuritic Aβ accumulations, not from cell body deposits. Certainly, it is possible that the endosomes in the cell body are later transported into the neurites, but this notion remains to be tested. Another interesting observation we made was that the neurites containing Aβ aggregates have fewer mitochondria, which cluster around the Aβ accumulations. Does this suggest a relationship between Aβ aggregation and disrupted/exhausted mitochondrial function? Future studies will tell. The CAD cell neuronal system proposed by us (4,6), as well as those used in the featured papers, SHSY5Y neuroblastoma cells (1,2), are ideal for pursuing such questions.
Amazingly, the accumulation of Aβ aggregates occurs in CAD cell cultures without any addition of Aβ to the medium. All Aβ is produced from the endogenous APP that the CAD cells normally express. We know that CAD cells actively produce Aβ, which polymerizes within the cells to form low and high oligomers, detectable with anti-Aβ and anti-oligomer antibodies. A detailed description of our view on how these neuritic Aβ accumulations could nucleate plaques in AD is given elsewhere (4,5).
Those interested could also take a look at our two SWAN-style hypotheses on the ARF website (6,7). To conclude, we have to sadly agree with Gunnar Gouras, who mentioned in his comment from 22 January 2010, the reluctance of many investigators to even consider the intracellular aspect of Aβ pathology. Researchers should definitely pay closer attention to the intracellular Aβ aggregates, because these could be key to the pathogenesis of AD.
See also:
Muresan, Z. and V. Muresan, CAD cells are a useful model for studies of APP cell biology and Alzheimer’s disease pathology, including accumulation of Aβ within neurites. SWAN Alzheimer Knowledge Base. Alzheimer Research Forum. 2009.
Muresan, Z. and V. Muresan, Brainstem Neurons Are Initiators of Neuritic Plaques. SWAN Alzheimer Knowledge Base. Alzheimer Research Forum.
References:
Friedrich RP, Tepper K, Rönicke R, Soom M, Westermann M, Reymann K, Kaether C, Fändrich M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A. 2010 Feb 2;107(5):1942-7. PubMed.
Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20324-9. PubMed.
Harper JD, Lansbury PT. Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385-407. PubMed.
Muresan Z, Muresan V. Neuritic deposits of amyloid-beta peptide in a subpopulation of central nervous system-derived neuronal cells. Mol Cell Biol. 2006 Jul;26(13):4982-97. PubMed.
Muresan Z, Muresan V. Seeding neuritic plaques from the distance: a possible role for brainstem neurons in the development of Alzheimer's disease pathology. Neurodegener Dis. 2008;5(3-4):250-3. Epub 2008 Mar 6 PubMed.
Make a Comment
To make a comment you must login or register.