Juvenile Lysosomes and Worn-Out Mitochondria Clog Axons in ALS Model
Quick Links
In amyotrophic lateral sclerosis, motor neurons struggle with axonal transport, autophagy, and mitochondrial maintenance. A July 15 paper in Neuron unites those three processes in a single theory: the mutant ALS protein SOD1 obstructs transport of endosomes, preventing them from maturing into lysosomes and from degrading worn-out mitochondria. Fixing the endosome transport gave mSOD1 model mice an extra 12 days of life, report Zu-Hang Sheng and colleagues at the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. Though Sheng conceded that may not be a huge chunk of time, he predicted that this treatment, added to one that boosts healthy mitochondria production, could do more.
Motor neurons from mSOD1 mice (right) make fewer mature lysosomes (green) than do neurons from wild-type mice (left). [Courtesy of Neuron, Xie et al.]
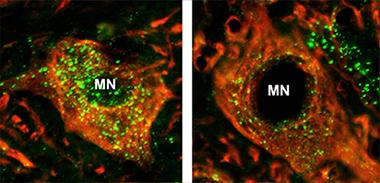
Lollygagging Lysosomes.
Mitochondria are malformed and poor powerhouses in people with ALS and in mouse models of the disease, stressing energy-guzzling motor neurons (see Aug 2014 news; news; Jan 2012 news). Part of the problem, researchers hypothesized, was that the organelles fester in axonal traffic jams (see Jan 2012 news; Nov 2009 conference news). Previously, Sheng’s lab tested this idea by crossing mSOD1 mice to a strain with heightened mitochondrial motility (see Jan 2008 news). However, freeing the mitochondria to move more made no difference to symptoms (Zhu and Sheng, 2011).
First authors Yuxiang Xie and Bing Zhou turned their attention to a different hypothesis: What if motor neurons cannot properly dispose of damaged, used-up mitochondria? Genes involved in autophagy, by which the cell disposes of unwanted organelles and vesicles, and mitophagy in particular already have been linked to ALS, suggesting defective waste management in ailing motor neurons (see Oct 2014 news; Feb 2015 news; Nov 2011 conference news). When Xie and Zhou examined spinal motor neurons from mSOD1 mice, they observed a dearth of mature lysosomes that began at 40 days and worsened with age (see image above). They also saw mitochondria stuck in autophagic vacuoles.
Autophagy does not work properly without efficient axonal transport. Garbage-toting endosomes must move toward the cell’s center, powered by a dynein motor protein, in order to mature into full-fledged lysosomes that can chew up proteins and organelles (Cai et al., 2010). This creates a particular challenge for motor neurons because they have lengthy axons, Sheng noted. Could poor transport explain the mitochondrial log jam? The authors cultured spinal motor neurons from adult mice and watched the way their endosomes moved. In neurons from mSOD1 mice, about one-quarter fewer endosomes travelled in the retrograde direction.
Snapin normally attaches endosomes to dynein motors (left), but mutant SOD1 gets in the way (right). [Courtesty of Neuron, Xie et al.]
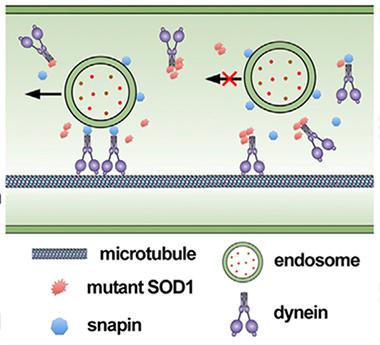
Running Interference.
The findings suggested that mutant SOD1 interfered with lysosomal transport and maturation, and Xie and Zhou had a good idea of how. Endosomes rely on an adapter protein, snapin, to latch them to the dynein motor, and other researchers discovered that mutant SOD1 binds the same spot on that motor (Zhang et al., 2007). Sheng and colleagues hypothesized that snapin and mSOD1 might compete for dynein (see image at left), and their data backed them up. They found that less dynein attached to endosomes from the spinal cords of mSOD1 mice than to wild-type endosomes. Co-immunoprecipitation experiments indicated that dynein and mSOD1 partnered, but adding snapin broke them up. Plus, when the authors added a snapin transgene to their mSOD1 neuron cultures, retrograde endosome transport improved, with about 20 percent more organelles moving toward the cell soma.
Finally, the authors used an adeno-associated virus to deliver snapin genes to the spinal cord of mSOD1 mice. When they examined the motor neurons of those mice, they saw the mitochondria looked healthier and held proper membrane potential. The treated animals maintained more spinal motor neurons, balanced better on a rotating cylinder, and lived for an average of 173 days, compared to 161 days for mice with normal snapin levels.
“We reveal, for the first time, progressive lysosomal deficits in the early stages of disease,” Sheng said. “Our study provides a new mechanism linking mutant SOD1 to impairment of retrograde transport to lysosome dysfunction, and finally to motor neuron death.”
Experts who spoke with Alzforum found the data convincing, but raised questions as well. “The rescue effect was minimal at only 12 days,” commented Philip Wong of the Johns Hopkins University School of Medicine in Baltimore, who was not involved in the study. “Even if [poor lysosomal transport] is an aspect of the pathogenesis, fixing it is not sufficient to fully attenuate disease—there must be other components at play here.” Giovanni Manfredi of Weill Cornell Medical College in New York noted that even if boosting lysosome maturation got rid of dysfunctional mitochondria, the neuron would still need fresh healthy ones. Sheng plans to address Manfredi’s concern by combining the snapin treatment with elevated mitochondrial biogenesis, hoping for more effective rescue. He also intends to examine lysosomal transport and maturation in other models, including mSOD1 mice with a slower-progressing disease, and motor neurons derived from the stem cells of people with the disease.
While Xie and Zhou discovered a way that mSOD1 could interfere with mitophagy, scientists speculated that the concept might be more general. Manfredi pointed out that dynein totes other cargoes besides lysosomes, and lysosomes degrade all kinds of cellular waste, so the trafficking defect could have broad consequences. In addition, Sheng said he has not ruled out transport defects in other organelles. Other disease-linked mutant proteins might also influence axonal transport, he added. His group will be examining the process in other mouse models of ALS genes as well as Parkinson’s and Huntington’s, he said.—Amber Dance
References
News Citations
- Mislocalized Mitochondria Sufficient for Motor Neuron Malaise
- Energy Crisis: ATP Deficiency Dooms Motor Neurons in Computer Model
- More Mitochondrial Mayhem in ALS Motor Neurons, Muscles?
- Mitochondria Stumble Their Way Along Axons in ALS Model
- Chicago: Axonal Transport Not So Fast in Neurodegenerative Disease
- Mitochondrial Gridlock—Syntaphilin Puts Brakes on Axonal Transport
- ALS, Parkinson’s Proteins Co-Mingle in Mitochondria Destruction Pathway
- TANK-Binding Kinase 1 Rumbles in as New ALS Gene
- DC: Protein Work Expands ALS/FTD Genetics
Paper Citations
- Zhu YB, Sheng ZH. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)-like disease in mutant SOD1 mice. J Biol Chem. 2011 Jul 1;286(26):23432-40. PubMed.
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron. 2010 Oct 6;68(1):73-86. PubMed.
- Zhang F, Ström AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J Biol Chem. 2007 Jun 1;282(22):16691-9. PubMed.
External Citations
Further Reading
Papers
- Kamat PK, Kalani A, Kyles P, Tyagi SC, Tyagi N. Autophagy of mitochondria: a promising therapeutic target for neurodegenerative disease. Cell Biochem Biophys. 2014 Nov;70(2):707-19. PubMed.
- Shi P, Wei Y, Zhang J, Gal J, Zhu H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;20 Suppl 2:S311-24. PubMed.
- Klinman E, Holzbaur EL. Stress-Induced CDK5 Activation Disrupts Axonal Transport via Lis1/Ndel1/Dynein. Cell Rep. 2015 Jul 21;12(3):462-73. Epub 2015 Jul 9 PubMed.
- Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci. 2011 May 25;31(21):7817-30. PubMed.
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012 Feb;8(2):108-17. PubMed.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013 Aug;19(8):983-97. PubMed.
News
- Sluggish Mitochondria Not Causing Withered ALS Axons After All?
- Paris: Intracellular Traffic and Neurodegenerative Disorders
- Slowly But Surely, Dynein Delivers Stress Signals in ALS Model
- Enhanced Autophagy Protects Against ALS Proteotoxicity
- Evidence Mounts That Mitochondrial Gene Is Bona Fide ALS, FTD Risk Factor
- Can Autophagy Protect ALS Cell Models from Mutant TDP-43?
- No MAM: ALS Protein Breaks Mitochondria-Endoplasmic Reticulum Bond
Primary Papers
- Xie Y, Zhou B, Lin MY, Wang S, Foust KD, Sheng ZH. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron. 2015 Jul 15;87(2):355-70. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.