No C9ORF72, No Problem: Knockout Mouse Neurologically OK
Quick Links
Mice missing both copies of the amyotrophic lateral sclerosis gene C9ORF72 balance and move just fine, and their neurons exhibit no signs of degeneration. The non-phenotype of the animals, reported in the June 5 Annals of Neurology online by researchers at the University Medical Center Utrecht in the Netherlands, makes an important point about how C9ORF72 hexanucleotide expansions might cause ALS in people who have only one normal copy of the gene. Researchers have theorized that either a gain of toxic function or a loss of normal C9ORF72 function could be the culprit. “Haploinsufficiency of C9ORF72, by itself, is insufficient to cause ALS,” concluded R. Jeroen Pasterkamp, co-senior author along with Leonard van den Berg.
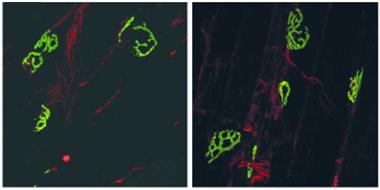
Neuromuscular Normal:
The neuron-muscle junctions of C9ORF72-knockout mice (right) appear nicely elaborated, just like wild-type (left). [Courtesy of Koppers et al., Ann. Neurol., John Wiley & Sons.]
First author Max Koppers and colleagues used floxed C9ORF72 and Nestin-Cre to eliminate the gene’s fourth and fifth exons from neurons and glia. They confirmed by PCR that the C9ORF72 RNA was reduced in the brain and spinal cord, and via western blotting that the protein was missing from the brain. The gene was expressed normally in other tissues. This differs from the status of human C9ORF72 expansion carriers, who express one normal copy in all cells, and thus produce low levels of C9ORF72 in the nervous system (Waite et al., 2014). The authors chose their approach so development would proceed normally until the Nestin promoter kicked on in 10-day-old embryos.
At 18 months old, the resulting mice exhibited entirely normal motor function, with no trouble balancing on a rotating rod or gripping a wire mesh. When the researchers examined their tissues, they observed the usual number and size of spinal motor neurons, no signs of gliosis, and healthy neuromuscular junctions. The animals lived a normal lifespan, with some lasting longer than two years. The only difference between the C9ORF72-negative mice and controls was a slightly reduced body weight, by a few grams, which the authors could not explain.
Koppers’ results differ from knockdown experiments in nematodes and zebrafish, where loss of C9ORF72 did lead to movement deficits (Ciura et al., 2013; Therrien et al., 2013). The discrepancy could arise, the authors suggest, because the worm and fish C9ORF72 genes are dissimilar to the human and mouse sequences. Authors of another study using antisense oligonucleotides to knock down C9ORF72 in mice observed no major side effects, backing up Kopper’s findings that loss of the gene makes little difference (see Nov 2013 news on Lagier-Tourenne et al., 2013).
A recently published mouse model supports the gain-of-function hypothesis, as well. Animals carrying the disease-linked expanded repeats exhibited neuropathology and symptoms reminiscent of frontotemporal dementia, which also results from C9ORF72 expansions (see May 2015 news).
Christine Vande Velde of the University of Montréal in Canada, who was not involved in the study, agreed that the sequence differences between mammal, fish, and worm C9ORF72 likely underlie the discrepant results from model organisms. She suggested it would be interesting to test for symptoms of dementia, as well as motor neuron disease, in the animals.—Amber Dance
References
News Citations
- Sense, Antisense: C9ORF72 Makes Both Forms of RNA, Peptides
- First C9ORF72 Mice Mimic Key Pathology, Behavior
Paper Citations
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014 Jul;35(7):1779.e5-1779.e13. Epub 2014 Jan 17 PubMed.
- Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol. 2013 May 30; PubMed.
- Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8(12):e83450. Epub 2013 Dec 12 PubMed.
- Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):E4530-9. Epub 2013 Oct 29 PubMed.
External Citations
Further Reading
Papers
- Panda SK, Wefers B, Ortiz O, Floss T, Schmid B, Haass C, Wurst W, Kühn R. Highly Efficient Targeted Mutagenesis in Mice Using TALENs. Genetics. 2013 Nov;195(3):703-13. PubMed.
- Mizielinska S, Isaacs AM. C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function?. Curr Opin Neurol. 2014 Oct;27(5):515-23. PubMed.
- Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A, Goate A, Ravits J, Baloh RH. Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging. 2013 Sep;34(9):2234.e13-9. PubMed.
- Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis-Dusenbery BN, Eggan K. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013 Dec;16(12):1725-7. Epub 2013 Nov 3 PubMed.
News
- Antisense RNA from C9ORF72 Repeats Is Likely Culprit in Patient Neurons
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
- C9ORF72’s Dirty Work Done by Problem Proteins
- C9ORF72 Killer Dipeptides Clog the Nucleolus
- RNA-DNA Pairs: At the Root of C9ORF72 Repeat Damage?
Primary Papers
- Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015 Sep;78(3):426-38. Epub 2015 Jul 3 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.