No Mere Microglial Marker, TMEM119 Supports Aβ Phagocytosis
Quick Links
The transmembrane protein 119 has a new job. Long known as a marker that distinguishes homeostatic microglia from macrophages and other myeloid cells, the protein also helps clear amyloid from the brain, according to scientists at the Chinese Academy of Science in Beijing led by Junliang Yuan, Yukai Wang, Zhao-Qian Teng, and Baoyang Hu. As they reported May 15 in Cell Immunity, the protein co-opts the cell surface receptor LRP1to facilitate Aβ phagocytosis. In so doing, TMEM119 seals its own fate, marking itself and Aβ for degradation. Boosting TMEM119 slowed memory loss in an AD mouse model.
- TMEM119, which supports microglial homeostasis, falters in amyloidosis mice.
- The protein promotes phagocytosis of Aβ, but is degraded along with it.
- Overexpressing TMEM119 stalled memory loss.
Renzo Mancuso of University of Antwerp in Belgium, who was not involved with the research, called the findings very interesting. The authors “provide compelling evidence that TMEM119—long considered a static marker of microglial identity—actually plays a pivotal and dynamic role in AD pathology,” he wrote to Alzforum (comment below).
First author Jing Liu and colleagues took an interest in TMEM119 because even though they found it bound Aβ in vitro, they noticed a sharp drop in levels of TMEM119 near amyloid plaques in mice. They sought to resolve this contradiction.
First, they looked at expression using single-nucleus multiomic sequencing. Because they found plaques did not affect transcript levels, they wondered whether microglia might be chewing up the transmembrane protein alongside amyloid. To investigate this, the authors focused on LRP1, known to transport extracellular molecules, including Aβ, into cells to be broken down by the endo-lysosomal system (Yang et al., 2025).
Liu found that TMEM119 and LRP1 rarely associated in wild-type but co-localized with Aβ in 5xFAD mice. Knocking out LRP1 in microglial cells in vitro quashed this complex, and it prevented TMEM119 disappearing from the cell surface when Aβ was added. The data indicated that microglia take up TMEM119 as they phagocytose Aβ peptides.
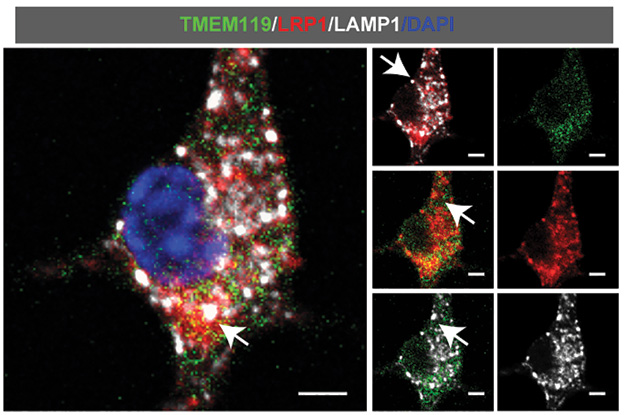
Microglial Magnet. In microglial cell cultures treated with Aβ, TMEM119 (green) co-localized with LRP1 (red) and the lysosomal marker LAMP1 (white). [Courtesy of Liu et al., 2025.]
How would dearth of TMEM119 affect microglial responses? To test this, the authors ran single-cell RNA sequencing on tissues from wild-type and 5xFAD mice, with and without TMEM119 knocked out, identifying gene expression profiles that characterized homeostatic, active, and transitional microglial states. In 5xFAD mice, TMEM119 deficiency provoked more microglia out of homeostasis and weakened phagocytosis. When Liu added Aβ40 or Aβ42 to microglia cultures, TMEM119 knockout cells took in less of the peptide than did wild-type cells, while microglia overexpressing TMEM119 took up more. Overexpressing TMEM119 also led to more Aβ40 and Aβ42 degradation, which the authors deduced was due to greater lysosomal function as evidenced by increased expression of the lysosomal marker, LAMP1.
The same scenario unfolded in vivo. In 5xFAD mice that overexpressed TMEM119, microglia surrounding plaques mustered more of the transmembrane protein and there were fewer plaques than in controls. The mice had less soluble and insoluble Aβ40 or Aβ42 in the cortex and they performed better on the Morris water maze and the Y maze.
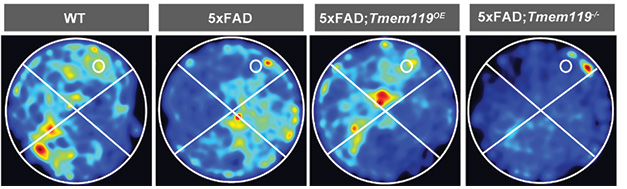
More TMEM119, Better Memory. In the Morris water maze, 5xFAD mice that overexpressed TMEM119 spent more time in the quadrant with the hidden platform (white circle). [Courtesy of Liu et al., 2025.]
“The findings pose the interesting idea that maintaining microglial homeostasis can be an avenue to slow down disease progression, as opposed to many current views and treatments that are aimed at activating microglia by agonizing ITAM receptors,” wrote Mancuso. Such agonists include antibodies to the cell-surface receptor TREM2, which helps clear amyloid from the brain (Apr 2025 news).
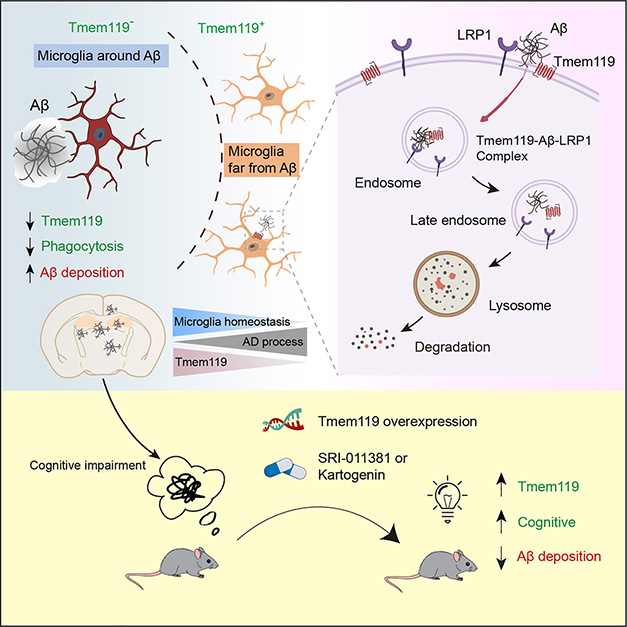
TMEM119 and Aβ. TMEM119 is diminished in microglia surrounding amyloid plaques, leading to less Aβ phagocytosis and less microglial homeostasis (top left). This is due in part to LRP1, which brings the Aβ and TMEM119 complex into the cell to be broken down via the endolysosomal system (top right). Reversing this decrease in TMEM119 through overexpression or drug treatment improved Aβ clearance and slowed cognitive decline in mice (bottom). [Courtesy of Liu et al., 2025.]
Indeed, the authors found two small molecules, kartogenin and SRI-011381, that mimicked TMEM119 overexpression. These are agonists of TGF-β1, a cytokine that promotes TMEM119 expression. Liu and colleagues fed 2-month-old 5xFAD mice with chow laced with either compound. Six months later, the mice had more microglia containing TMEM119, and the phagocytic marker CD68, than did untreated animals. Plaque burden was lower in the cortex and hippocampus and the mice performed better on the two maze tests. The scientists repeated the treatment in two other sets of mice: 5-month-old 5xFAD mice, which have a heftier plaque load, and 2-month-old female 3xTG mice. At 8 months, the 5XFAD mice had fewer plaques than controls, and memory and spatial recognition were similar to those of wild-type mice. At 6 months, the 3xTG mice remembered as well as did controls.
Hu and colleagues hope to determine whether homeostatic or activated microglia more effectively limit AD pathology. This remains an unresolved question for the field. For his part, Hu thinks targeting TMEM119 could be key to optimizing microglial action. “This dynamic process holds promise for opening new therapeutic avenues in Alzheimer’s disease,” he said.—Lauren Schneider
Lauren Schneider is a freelance writer in New York City.
References
Research Models Citations
News Citations
Paper Citations
- Yang W, Wei Z, Wang T. Unraveling the Role of LRP1 in Alzheimer's Disease: A Focus on Aβ Clearance and the Liver-Brain Axis. J Mol Neurosci. 2025 Apr 1;75(2):43. PubMed.
Further Reading
Papers
- Ruan C, Elyaman W. A New Understanding of TMEM119 as a Marker of Microglia. Front Cell Neurosci. 2022;16:902372. Epub 2022 Jun 13 PubMed.
Primary Papers
- Liu J, Wang Z, Liang W, Zhang Z, Deng Y, Chen X, Hou Z, Xie Y, Wang Q, Li Y, Bai C, Li D, Mo F, Wang H, Wang D, Yuan J, Wang Y, Teng ZQ, Hu B. Microglial TMEM119 binds to amyloid-β to promote its clearance in an Aβ-depositing mouse model of Alzheimer's disease. Immunity. 2025 May 9; Epub 2025 May 9 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
VIB-Center for Molecular Neurology
In this very interesting piece of work, Liu et al. provide compelling evidence that TMEM119—long considered a static marker of microglial identity—actually plays a pivotal and dynamic role in AD) pathology. It appears that TMEM119 can bind directly to small, soluble Aβ species, which subsequently leads to the recruitment of the receptor LRP1 to form a degradation complex, enabling Aβ clearance through lysosomal pathways. However, this clearance comes at a cost: The process leads to TMEM119 degradation itself, creating a paradox in which microglia lose the very tool they need to keep removing Aβ.
The importance of TMEM119 for Aβ clearance is nicely illustrated by the fact that, whereas its deficiency accelerates microglial transition into a dysfunctional, disease-associated DAM state, worsening Aβ buildup and cognitive decline, overexpressing TMEM119 restores microglial balance, boosts Aβ phagocytosis, reduces plaque burden, and significantly improves cognitive performance.
I believe the findings presented here have three key important conceptual implications. First, they challenge our view of TMEM119 as being just a cell-state marker; rather, TMEM119 appears to be a functional regulator of neuroimmune activity. This would offer new therapeutic angles beyond targeting Aβ itself.
Second, the findings pose the interesting idea that maintaining microglial homeostasis can be an avenue to slow down disease progression, as opposed to many current views and treatments that are aimed at activating microglia by agonizing ITAM receptors. Finally, this study reinforces the notion that timing is a key factor, because strategies that might work in early or mid-stage AD may not be effective later, underscoring the need for stage-specific therapeutic approaches.
Altogether, although there have been many attempts before at reducing microglial activation and restoring homeostasis as a potential therapeutic avenue for AD, this work represents a major contribution as it provides detailed molecular pathways that can be harnessed to restore microglial homeostasis and potentially modify disease.
Mayo Clinic
This is an interesting paper, investigating the role of TMEM119 in Alzheimer’s disease. While TMEM119 has been known as a specific marker of homeostatic microglia, the authors demonstrated that TMEM119 mediates phagocytosis of Aβ by forming a complex with Aβ and LRP1. As LRP1 interacts with various ligands related to AD, including tau, APOE, and C1q, it would be important to define how TMEM119 contributes to the phagocytosis of such ligands.
Since the authors showed that TMEM119 deficiency accelerates the transition of microglia from a homeostatic state to stage 1 disease-associated microglia (DAM) in mouse models of amyloidosis, LRP1 deficiency may do the same. It might be interesting to investigate if LRP1 deficiency influences the transition of stage 1 DAM to stage 2.
Of note, the authors found that TMEM119 overexpression ameliorated amyloid pathology in mice. While increased TMEM119 levels may directly facilitate Aβ phagocytosis by microglia, it may also swell the homeostatic microglial population, influencing Aβ clearance regardless of TMEM119-Aβ binding. Future studies should define how homeostatic microglia mediate Aβ phagocytosis and whether TMEM119 overexpression induces the transition to specific types of homeostatic microglia.
Make a Comment
To make a comment you must login or register.