Paper Alert: Autoimmunity in Another C9ORF72-Deficient Mouse Strain
Quick Links
Hexanucleotide expansions in an intron of C9ORF72 are linked to amyotrophic lateral sclerosis and frontotemporal dementia, but how these expansions trigger disease is largely a mystery. As described in Science Translational Medicine on July 13, yet another of several C9ORF72-deficient mouse strains points to immune dysfunction as a potential player. Although two other recently described C9-deficient mouse strains developed inflammation, the newly described strain developed full-fledged autoimmunity and died young.
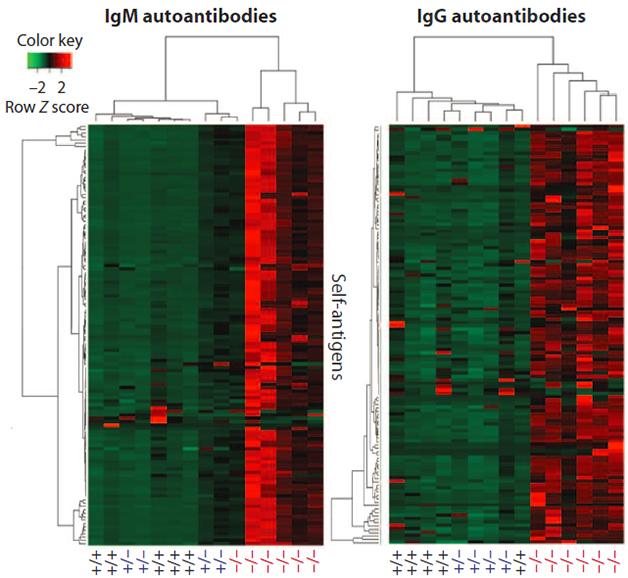
Autoimmune Profile. Plasma autoantibody levels reveal C9ORF72-homozygous knockouts (-/-), and some of the heterozygous (+/-) animals display a similar autoimmune profile for IgM autoantibodies, though not IgG. [Image courtesy of Burberry et al., Science Translational Medicine 2016.]
Even though immune dysfunction is not equally severe in recent C9ORF72 knockout strains, the commonalities are greater than the differences, and they send a clear message, said senior author Kevin Eggan of Harvard University. “This protein is a potent suppressor of inflammation and autoimmunity,” he told Alzforum.
Eggan presented the bulk of the results at the 10th Brain Research Conference: RNA Metabolism in Neurological Disease, in Chicago last October (see Oct 2015 conference series). The paper delves further into the extent of autoimmunity detected in the mice.
First author Aaron Burberry and colleagues suppressed expression of C9ORF72 by replacing exons 2-6 with a LacZ and neomycin resistance cassette. They dubbed these mice “KOMP” because the initial gene targeting was performed by the National Institutes of Health Knock Out Mouse Project, which aims to develop knockout strains of every gene in the mouse genome. Robert Baloh’s group at Cedars-Sinai Medical Center in Los Angeles generated a recently published C9ORF72 knockout strain in the same way (see Mar 2016 news on O’Rourke et al., 2016). In addition to the KOMP strain, Eggan’s group developed a strain lacking the neomycin resistance cassette because they were concerned that the insert may foul up expression of nearby genes. This “neo-deleted” strain was generated in outbred mice, as opposed to the KOMP strain, which was on a C57BL/6 background.
As presented in Chicago, the knockout mice developed severe inflammation. Their blood swarmed with inflammatory cytokines, their spleens were grossly enlarged. T cells, B cells, and neutrophils overpopulated both the spleen and blood. Interestingly, heterozygous KOMP mice were normal on all these measures.
Both the C9ORF72 homozygous and—surprisingly, perhaps—heterozygous knockout mice died young. Compared with wild-type animals, 98 percent of which lived at least 400 days, KOMP+/- and KOMP-/- mice started perishing around 70 days, with just 69 percent and 7 percent surviving until the 400-day mark, respectively. The corresponding neo-deleted strains lived longer than the KOMP strains, with 76 percent of heterozygotes and 64 percent of homozygotes surviving until 400 days. Necropsy revealed several potential causes of death, including respiratory failure, internal hemorrhage, severe ataxia, lymphocytic stromal cell hyperplasia, and enlarged livers.
The researchers attributed the inflammatory phenotype and early death to hematopoietic cells lacking C9ORF72 based on a series of transplantation experiments. Bone marrow from knockout mice into wild-type mice that had been irradiated to destroy their hematopoietic system increased inflammation and shortened the recipients’ lifespans, relative to irradiated wild-type recipients that received wild-type bone marrow. The reverse transplantation—putting wild-type bone marrow into irradiated KOMP mice—reduced inflammation and extended life.
Given the proliferation of B and T cells in the knockouts, the researchers wondered if autoimmunity was responsible for the inflammatory phenotype, and for the animals’ early demise. To test this, Burberry looked for antibodies to auto antigens. He found antibodies that bound double-stranded DNA in the plasma of homozygous but not heterozygous knockouts. These findings dovetail with those from the C9ORF72 knockout mouse developed by Venus Lai’s group at Regeneron in Tarrytown, New York. Serum from those mice contained antibodies directed against small ribonuclear proteins called Smiths. These autoantibodies are often found in people with systemic lupus erythematosus. Like people with lupus, Lai’s mice also had kidney damage (see Mar 2016 news on Atanasio et al., 2016).
The presence of anti-dsDNA antibodies is a known sign of autoimmunity, but can also indicate rapid cell death due to other causes. To distinguish between these possibilities, Burberry screened the plasma using an autoantigen microarray, which contains snippets of 124 autoantigens from various cell types. Compared to wild-type mice, homozygous C9ORF72 knockouts had elevated levels of IgM antibodies against 117 of these antigens, and IgG antibodies against 113. In heterozygous mice, on the other hand, IgM antibodies against one and IgG antibodies against just two of the antigens surpassed wild-type levels, though others trended higher.
Hierarchical cluster analysis of the autoantibody response as a whole indicated that one in two heterozygous mice had a similar pattern of IgM autoantibody responses to that of the homozygous mice. The study used only four heterozygous and six homozygous knockouts. Still, to Eggan, these similarities point to an inkling of autoimmunity in mice with a single functional copy of C9ORF72, a genotype that would be expected to better model disease in people who are heterozygous for C9ORF72 expansions. “This experiment hit the most pay dirt with respect to whether there is some kind of intermediate phenotype in heterozygous animals,” Eggan said.
The authors claim their knockout mice bear a striking resemblance to mice lacking expression of Tbk1, a kinase involved in IFN-γ signaling and autophagy. Loss-of-function mutations in the TBK1 underlie rare forms of familial ALS and FTD (see Feb 2015 news; Mar 2015 news). Tbk1 knockouts develop enlarged spleens, and their immune cells infiltrate the skin, lung, liver, and kidneys. Whether C9ORF72 and Tbk1 work through a common pathway to precipitate disease is unknown.
Do these findings make ALS an autoimmune disease? That remains to be seen. Immune cells do infiltrate the spinal cord and motor cortex in ALS patients and animal models of the disease, but researchers are conflicted on whether those immune cells are good or bad for motor neurons (see Sep 2009 news; Henkel et al., 2004; Troost et al., 1989). Burberry and colleagues acknowledge that while epidemiological evidence suggests a higher incidence of autoimmunity among people who develop ALS (Turner et al., 2013), evidence for autoantibodies in ALS patients is contradictory (see Zhao et al., 2013; Pagani et al., 2011).
What does the autoimmune phenotype say about ALS in C9ORF72 expansion carriers? While some of the mice had weak hind limbs and lost motor control, they only partially recapitulated the phenotype of ALS, nor did they harbor overt neuropathology. Both Eggan and commentators proposed that a combination of loss- and gain-of-function mechanisms could be at play in expansion carriers. The expansions lead to RNA foci and dipeptide repeat inclusions in neurons (gain of function), while the reduced C9 expression simultaneously impairs immune cells from cleaning up the mess (loss of function). “A combination of these things leads to disease,” Baloh said. “At this point, to ignore one or the other is not a good idea. There’s evidence for both.”
To investigate whether a combination of loss- and gain-of-function mechanisms conspire to trigger disease, researchers are developing mouse strains that express a normal copy and an expanded copy of C9ORF72, just as humans do. Mice expressing the expansions (along with two normal copies of C9ORF72) already exist, and while they develop RNA foci and dipeptide repeats, they differ in the extent of their neurodegeneration or ALS-like symptoms (see C9-BACexp (Baloh/Lutz model) mice; C9-BAC500 (Brown model) mice; Dec 2015 news; May 2016 news).
“Given the variability in the human disease, perhaps it is not surprising that a picture of multiple causal factors and modifiers is being uncovered,” commented Johnathan Cooper-Knock of the University of Sheffield (see full comment below).
Based on the autoimmune phenotype his mice displayed, Eggan cautioned against developing therapeutic approaches that could knock down expression of the normal copy of C9ORF72. He’d rather target only the expanded allele. Furthermore, the findings may help clinicians identify inflammatory markers that could be used to monitor patients in clinical trials.—Jessica Shugart
References
News Citations
- C9ORF72 Knockout Causes Inflammation, not Neurodegeneration
- TANK-Binding Kinase 1 Rumbles in as New ALS Gene
- Second Study Salutes TANK-Binding Kinase 1 as ALS Gene
- ALS: T Cells Step Up
- C9ORF72 Mice A-OK Despite Toxic RNAs, Peptides
- New C9ORF72 Mice Develop Symptoms Resembling ALS/FTD
Research Models Citations
Paper Citations
- O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016 Mar 18;351(6279):1324-9. PubMed.
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016 Mar 16;6:23204. PubMed.
- Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004 Feb;55(2):221-35. PubMed.
- Troost D, van den Oord JJ, de Jong JM, Swaab DF. Lymphocytic infiltration in the spinal cord of patients with amyotrophic lateral sclerosis. Clin Neuropathol. 1989 Nov-Dec;8(6):289-94. PubMed.
- Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology. 2013 Oct 1;81(14):1222-5. Epub 2013 Aug 14 PubMed.
- Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013 Sep;8(4):888-99. Epub 2013 Jul 25 PubMed.
- Pagani MR, Gonzalez LE, Uchitel OD. Autoimmunity in amyotrophic lateral sclerosis: past and present. Neurol Res Int. 2011;2011:497080. PubMed.
Other Citations
Further Reading
No Available Further Reading
Primary Papers
- Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, Koszka K, Li QZ, Zon L, Rossi DJ, Trowbridge JJ, Notarangelo LD, Eggan K. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016 Jul 13;8(347):347ra93. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Sheffield
This detailed study from Burberry et al. describes a new mouse model with C9ORF72 loss-of-function. The study largely confirms the findings of earlier studies (O’Rourke et al., 2016; Atanasio et al., 2016)—loss of the murine C9ORF72 ortholog results in splenomegaly and proliferation of immune cells without a notable neuromuscular phenotype. Like the Atanasio et al. study, Burberry and colleagues identified an autoimmune phenotype. Interestingly they showed that transplantation of wild-type bone marrow was sufficient to ameliorate the phenotype of C9ORF72-null mice.
Whilst this has clear implications for proposed antisense oligonucleotide therapy aimed at reducing expression of mutant C9ORF72, the authors also draw attention to wider evidence for immune dysfunction in amyotrophic lateral sclerosis (ALS) and in particular, the striking similarity between the phenotype of these mice and TBK1-mull mice (Marchlik et al., 2010). The suggestion is that C9ORF72 loss-of-function may be important to ALS pathogenesis even if it is not the primary cause of neurotoxicity.
Indeed the role of neuroinflammation in neurodegenerative disease is a growing area; an attractive hypothesis is that patients with heterozygous mutations in C9ORF72 experience immune dysfunction due to haploinsufficiency, which interacts synergistically with a gain of function of the GGGGCC-repeat expansion, resulting in neuronal-specific toxicity. We await studies describing C9ORF72-null mice engineered to express the expanded human gene. However, a recent study from the lab of Laura Ranum suggested that expression of the expansion in the context of the full-length human C9ORF72 gene is sufficient to recapitulate a motor neuron disease phenotype (Liu et al., 2016). Given the variability in the human disease, perhaps it is not surprising that a picture of multiple causal factors and modifiers is being uncovered.
References:
O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016 Mar 18;351(6279):1324-9. PubMed.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016 Mar 16;6:23204. PubMed.
Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, Goutagny N, Kuang W, Askew GR, Roberts V, Benoit S, Zhou T, Ling V, Pfeifer R, Stedman N, Fitzgerald KA, Lin LL, Hall JP. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol. 2010 Dec;88(6):1171-80. Epub 2010 Jul 22 PubMed.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016 May 4;90(3):521-34. Epub 2016 Apr 21 PubMed.
Make a Comment
To make a comment you must login or register.