C9ORF72 Knockout Causes Inflammation, not Neurodegeneration
Quick Links
Three new mouse lines that scientists expected to model aspects of amyotrophic lateral sclerosis and frontotemporal dementia have no neural problems. They do have enlarged spleens and lymph nodes. Macrophages and microglia were defective and inflammation appeared rampant in the animals, which lacked one or two copies of the murine version of the C9ORF72 gene. Loss of C9ORF72 alone does not lead to neurodegeneration, the researchers concluded. One report suggests the mice model the autoimmune disorder lupus.
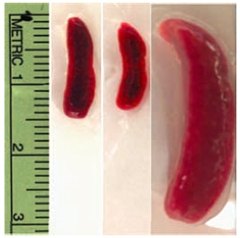
Outsize organs.
The spleens of C9ORF72 knockout mice (right) are much larger than average (left/middle). [Courtesy of Science/AAAS.]
Hexanucleotide expansions in an intron of the C9ORF72 gene cause ALS or FTD (see Sep 2011 news). Scientists do not know precisely what the C9ORF72 gene product does normally, though its structure suggests it manages membrane events, such as fusions (see Jan 2013 news). Scientists are also working to discover whether the loss of one normal C9ORF72 allele, or the production of toxic RNAs and peptides from the repeat sequence, causes neurodegeneration. Mice lacking the gene in just their nervous systems appear to be fine (see Jun 2015 news; Nov 2013 news). The new papers describe mice lacking C9ORF72 in the entire genome.
In the March 18 Science, collaborators from Cedars-Sinai Medical Center in Los Angeles and The Jackson Laboratory (JAX) in Bar Harbor, Maine, describe two of the new mice. Researchers led by Robert Baloh at Cedars-Sinai characterized mice missing exons 2-6 of the C9ORF72 gene. They obtained the mice from the National Institutes of Health’s Knockout Mouse Project, a resource that aims to generate knockouts of every gene in the mouse genome. The KOMP C9ORF72 knockouts were made in a similar way to a separate knockout line lacking exons 2-6, studied by Kevin Eggan of Harvard University, and a previous line characterized by Don Cleveland of the University of California, San Diego, and Clotilde Lagier-Tourenne of Massachusetts General Hospital in Charlestown (see Oct 2015 conference news and table). Because exon 2 contains the start codon for the protein, none of these mice make C9ORF72. In the same Science paper, collaborator Cat Lutz and colleagues at JAX report their line of mice called F12, which lacks 73 base pairs around the gene’s start codon and make no protein either.
The Lutz and Baloh mice had similar phenotypes, supporting the idea that the C9ORF72 deletion was behind those changes. Further support comes from a third mouse line, published in the March 16 Scientific Reports, in which senior author Venus Lai and colleagues at Regeneron Pharmaceuticals in Tarrytown, New Jersey, excised the whole gene. Neither group knew that the other was working on the same problem, but all were reassured to discover they had similar results.
Overall, mice missing C9ORF72 developed normally. They showed no sign of motor neuron disease, balancing on a rotating rod as well as controls. In Science, first author Jacqueline O’Rourke and colleagues report that KOMP mice as old as 17 months had normal numbers of neurons in the brain and spinal cord. Both KOMP and F12 mice lived normal lifespans. The Regeneron full knockouts fared poorly, however, with eight out of 17 animals dying before 60 weeks, when the scientists sacrificed the rest for study.
Both the KOMP and Regeneron mice were sickly. In open-field tests, they roamed less than controls, whereas the F12 mice explored normally. It was readily apparent why the mice might not feel like strolling. Their spleens and lymph nodes were huge (see image), filled with large, debris-laden myeloid cells, including macrophages.
In this regard, these mice are similar to C9ORF72 nulls reported by the Eggan and Cleveland/Lagier-Tourenne groups, but the new studies offer more detail on the specific role C9ORF72 plays in the immune system.
Intracellular Issues
O’Rourke and co-authors focused on the cellular mechanisms behind the altered myeloid cells. She found that the swollen myeloid cells in the KOMP mice contained abnormally high levels of autophagy markers p62 and LC3, indicating defects in trafficking from endosomes to lysosomes. This would create a problem for macrophages gobbling up pathogens and infected cells, and could lead to inflammation, the authors predicted.
To study the importance of C9ORF72 in macrophages directly, O’Rourke and co-authors isolated bone marrow stem cells from their C9ORF72-null mice and differentiated them into macrophages. Late endosomes and lysosomes accumulated in these cells, indicating stalled traffic in the autophagy pathway. Transducing the cells with the C9ORF72 gene reversed the lysosome accumulation, confirming the lack of C9ORF72 had created the defect.
What about in the brain? In the nervous system, microglia assume macrophage roles, so O’Rourke and colleagues wondered if they, too, were altered in the absence of C9ORF72. In perusing published records of gene expression in the brain, Baloh was surprised to learn that microglia typically express more C9ORF72 than neurons, suggesting they might be particularly sensitive to its loss (Zhang et al., 2014; Sharma et al., 2015; Butovsky et al., 2014).
Indeed, just like the macrophages, microglia from the null mice accumulated endosomes and lysosomes. They also produced high levels of the inflammatory cytokines IL-6 and IL-1b. To find additional evidence of inflammation, the authors compared gene expression profiles among spinal cords from 17-month-old C9ORF72-null and control mice. Of 19 pathways upregulated in the null mice, nearly one-third had to do with inflammation. Despite these changes, the brains of all C9ORF72-null animals appeared normal.
Does the microglial/inflammatory phenotype correspond to any feature of ALS? To find out, O’Rourke and colleagues cross-referenced the mouse microglial expression profiles with gene expression data from people with sporadic or C9ORF72-based ALS (see Jul 2015 news). Of the six pro-inflammatory pathways upregulated in the C9ORF72-null mice, five were also upregulated in the brains of C9ORF72 expansion carriers, whereas only one was upregulated in sporadic ALS cases. Microglia in the motor cortices and spinal cord tissues from three people who died of C9ORF72-based ALS were also full of lysosomes.
“It is a very convincing argument that C9ORF72 is an essential gene to cells of myeloid origin,” commented Christine Vande Velde of the University of Montréal, who did not participate in the work. “It may be that there is a differential effect of the C9ORF72 expansion in the microglia cells compared to the neuronal cells.” The neurons seem to suffer from the repetitive RNAs and peptides produced by the expansion, she and Baloh agreed.
“C9ORF72 loss of function would not be expected to produce neural toxicity in isolation, but might by working synergistically with other mechanisms,” commented Johnathan Cooper-Knock of the University of Sheffield in the United Kingdom, who was not involved in either paper. For example, O’Rourke and colleagues suggest defective microglia might fail to clear aggregates from the nervous system and that could eventually cause trouble for neurons. However, Cooper-Knock was not ready to blame all the inflammation in the mice on problems in the myeloid cell lineage alone. To prove that would require a demonstration that mice with C9ORF72 missing only from myeloid cells have the same illness (see full comment below).
Baloh plans to create models that lack C9ORF72 specifically in microglia. He also intends to cross the null mice with a line overexpressing expanded C9ORF72 to determine if mice carrying the mutant allele but no normal C9ORF72 get sicker than the parent strains (see Dec 2015 news).
Auto-Immunity
At Regeneron, Lai and first author Amanda Atanasio called in co-author Vilma Decman and other colleagues versed in immunology to help them understand the C9ORF72-null phenotype. The mice possessed more circulating immune cells, including neutrophils, monocytes, and eosinophils, than controls did, and their blood concentration of certain cytokines and chemokines was higher. RNA sequencing found expression differences in immune response and inflammation genes. These features all point toward systemic inflammation and likely explain why the mice were sluggish during open-field tests, commented co-author Susannah Brydges. “If you or I had such massive expansion of immune cells and circulating cytokines—we would be feeling sick and be in bed,” she said.
Decman and colleagues found a proliferation of antibody-producing plasma cells in the spleens and lymph nodes of the null mice. IgM and IgG antibody classes were elevated in the serum. Proliferating plasma cells are often associated with autoimmunity, and sure enough, the C9ORF72-null mouse serum contained autoantibodies against the core proteins, called Smiths, of small ribonuclear proteins. These kinds of antibodies would make the mice ill, commented Brydges.
Those anti-Smith antibodies suggested the mice might mimic systemic lupus erythematosus. In people, this autoimmune disease attacks a variety of tissues, including the skin and joints. Symptoms include joint pain and swelling, hair loss, fever, and sensitivity to sunlight. A majority of people who possess anti-Smith antibodies come down with lupus, said Brydges. Supporting the hypothesis, the C9-null mice also sustained kidney damage, a common problem in people with lupus. The Regeneron scientists are now searching for C9ORF72 mutations or expansions in people diagnosed with this autoimmune disease.
People with ALS are not known to have systemic immune abnormalities, Baloh said—but then, physicians have not had reason to check. Both he and Atanasio suggested doctors might want to take an infection history and assess immune symptoms more closely in ALS patients. The literature contains at least one other hint of a relationship between C9ORF72 and autoimmunity. Cooper-Knock and colleagues reported that in those rare, unfortunate instances where ALS and the autoimmune disorder multiple sclerosis coincide, patients are likely to carry C9ORF72 expansions (Ismail et al., 2013).
“These papers are important because they shed light on the normal role of C9ORF72 in lysosomal function, cytokine-mediated inflammation, and immune response,” commented Ronald Klein of the Louisiana State University Health Sciences Center in Shreveport, who did not participate in either project.
Both papers suggest that while immune function requires C9ORF72, its loss creates no ALS phenotype, Vande Velde said. Neither group analyzed FTD-like phenotypes in detail, so the exact relationship between C9ORF72 loss of function and frontotemporal lobe degeneration remains uncertain, she added.
To O’Rourke, the mice raise a red flag for scientists aiming to quell ALS with antisense oligonucleotides (ASOs) against C9ORF72 (Oct 2013 news; Jan 2013 conference news). The body’s macrophages and microglia need some C9ORF72 to function properly, so it might be wise to dampen only the expanded allele.
Researchers have come up with expansion-specific ASOs, and anticipate starting human trials within a year or two (see Nov 2015 conference news). These studies should monitor for inflammation, said Vande Velde.—Amber Dance
References
News Citations
- Corrupt Code: DNA Repeats Are Common Cause for ALS and FTD
- C9ORF72 Function: Is the ALS Protein a Membrane Traffic Cop?
- No C9ORF72, No Problem: Knockout Mouse Neurologically OK
- Sense, Antisense: C9ORF72 Makes Both Forms of RNA, Peptides
- C9ORF72 Mice Point to Gain of Toxic Function in ALS, FTD
- C9ORF72, Sporadic ALS Transcriptomes Now Available
- C9ORF72 Mice A-OK Despite Toxic RNAs, Peptides
- Second Study Confirms Antisense Oligonucleotides Bust RNA Aggregates
- Chicago—RNA Inclusions Offer Therapeutic Target in ALS
- Listen Up, Gene Silencing Strikes a Chord at RNA Meeting
Paper Citations
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014 Sep 3;34(36):11929-47. PubMed.
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015 Dec;18(12):1819-31. Epub 2015 Nov 2 PubMed.
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014 Jan;17(1):131-43. Epub 2013 Dec 8 PubMed.
- Ismail A, Cooper-Knock J, Highley JR, Milano A, Kirby J, Goodall E, Lowe J, Scott I, Constantinescu CS, Walters SJ, Price S, McDermott CJ, Sawcer S, Compston DA, Sharrack B, Shaw PJ. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J Neurol Neurosurg Psychiatry. 2013 Jan;84(1):79-87. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol. 2013 May 30; PubMed.
- Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8(12):e83450. Epub 2013 Dec 12 PubMed.
News
- C9ORF72 Mice A-OK Despite Toxic RNAs, Peptides
- First C9ORF72 Mice Mimic Key Pathology, Behavior
- RNA Regulator Locked Out of Nucleus by C9ORF72 Repeats
- C9ORF72 RNA Foci Acquitted of Toxic Charge—in Fruit Flies
- ALS Gene Repeats Obstruct Traffic Between Nucleus and Cytoplasm
- Antisense RNA from C9ORF72 Repeats Is Likely Culprit in Patient Neurons
Primary Papers
- O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016 Mar 18;351(6279):1324-9. PubMed.
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016 Mar 16;6:23204. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Sheffield
This study from the Baloh group describes the generation and characterization of mice deficient in the C9orf72 ortholog 3110043O21Rik. O’Rourke and colleagues generated heterozygous and homozygous C9orf72-null mice by two independent methods. Dose-dependent reduction in levels of the C9orf72 ortholog is demonstrated in heterozygous and homozygous mice obtained from both strategies. None of the mice developed any neuromuscular dysfunction, but both lines displayed splenomegaly and lymphadenopathy, which the authors hypothesize was related to dysfunction of myeloid cells, including peripheral macrophages and CNS microglia. In isolated myeloid cells they demonstrate accumulation of Lamp1-positive material, suggesting a deficiency in autophagy pathways; this is accompanied by a pro-inflammatory cytokine response and is rescued by the expression of human C9orf72.
Autophagy dysfunction is consistent with what is already known about C9orf72 function: Analysis of C9orf72 protein domains led to the hypothesis that it is a DENN-domain protein with a role in vesicle trafficking (Levine et al., 2013). This was supported by the observed sign epistasis with TMEM106B (Gallagher et al., 2014, reviewed in Cooper-Knock et al., 2015). At the ALS Symposium in December 2015, Clotilde Lagier-Tourenne reported early findings from another loss-of-function C9orf72 mouse model that had been interpreted as signs of lymphoma. While we await the final paper, many of the changes she described are similar to those outlined in this report, suggesting the two studies might be consistent.
Atanasio and colleagues report a similar phenotype with splenomegaly, lymphadenopathy, and proliferation of myeloid cells. In fact, this mouse model has perhaps a more severe phenotype, because homozygous C9orf72 nulls developed an autoimmune syndrome with similarities to systemic lupus erythematosus, including prominent glomerulonephropathy and even motor weakness, but not a classical ALS-like phenotype.
The strength of this report from O'Rourke et al. lies in the consistency of the findings from two mouse lines. The suggestion that all of the inflammatory changes seen are secondary to the dysfunction of the myeloid cells is interesting but not proven beyond all doubt in this study; that would require demonstration that the myeloid cell abnormalities are necessary and sufficient to produce the entire phenotype.
O'Rourke et al. relate their findings to human C9orf72 disease by demonstrating accumulation of Lamp1-positive material in microglia of C9orf72-ALS patients and observing similar transcriptome changes (particularly with respect to genes involved in inflammation) in the mice to those seen in a study of C9orf72-ALS CNS tissue (Prudencio et al., 2015). This leads to an attractive hypothesis: Loss of C9orf72 function contributes to disease pathogenesis via impaired clearance of protein aggregates by microglia, but the aggregates might develop in response to other stimuli. Therefore C9orf72 loss of function would not be expected to produce neuronal toxicity in isolation, but might by working synergistically with other mechanisms. In the context of C9orf72 disease, a number of toxic gain-of-function pathways have been proposed based on RNA foci and dipeptide repeat proteins derived from the C9orf72 GGGGCC-repeat expansion. The authors point to studies suggesting a loss of C9orf72 function in the human C9orf72 disease and studies linking ALS and microglial dysfunction as support for their hypothesis. However, without a neurodegenerative phenotype in the C9orf72-null mice, this remains speculation to some extent. Indeed, Atanasio et al. come to the opposite conclusion despite very similar observations, though it should be noted that Atanasio and colleagues did not specifically compare their findings to the human disease or assess autophagy in C9orf72-null myeloid cells.
It will be fascinating to see what happens when these C9orf72-null mice are crossed with the C9BAC mice recently reported by the same authors (O’Rourke et al., 2015). The C9BAC mice express the GGGGCC-expansion in the context of the full length human C9orf72 gene via a bacterial artificial chromosome (BAC); they develop RNA foci and dipeptide repeat protein inclusions but not other important features of C9orf72 disease, including neurodegeneration and, notably, neuroinflammation. If the hypothesis proposed by O’Rourke and colleagues is correct, then combining the two models might finally precipitate neuronal toxicity.
References:
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013 Feb 15;29(4):499-503. Epub 2013 Jan 16 PubMed.
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer JD, Halliday G, Van Broeckhoven C, Seilhean D, Shaw PJ, Frosch MP, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns NJ, Cooper-Knock J, Cotman C, Cras P, Cruts M, De Deyn PP, DeCarli C, Dobson-Stone C, Engelborghs S, Fox N, Galasko D, Gearing M, Gijselinck I, Grafman J, Hartikainen P, Hatanpaa KJ, Highley JR, Hodges J, Hulette C, Ince PG, Jin LW, Kirby J, Kofler J, Kril J, Kwok JB, Levey A, Lieberman A, Llado A, Martin JJ, Masliah E, McDermott CJ, McKee A, McLean C, Mead S, Miller CA, Miller J, Munoz DG, Murrell J, Paulson H, Piguet O, Rossor M, Sanchez-Valle R, Sano M, Schneider J, Silbert LC, Spina S, van der Zee J, Van Langenhove T, Warren J, Wharton SB, White CL 3rd, Woltjer RL, Trojanowski JQ, Lee VM, Van Deerlin V, Chen-Plotkin AS. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014 Mar;127(3):407-18. Epub 2014 Jan 19 PubMed.
Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, Shaw PJ. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS One. 2015;10(5):e0127376. Epub 2015 May 27 PubMed.
Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P, Bieniek KF, DeTure M, Lee WC, Biendarra SM, Davis MD, Baker MC, Perkerson RB, van Blitterswijk M, Stetler CT, Rademakers R, Link CD, Dickson DW, Boylan KB, Li H, Petrucelli L. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci. 2015 Aug;18(8):1175-82. Epub 2015 Jul 20 PubMed.
O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L, Lee EB, Lutz CM, Baloh RH. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015 Dec 2;88(5):892-901. PubMed.
Make a Comment
To make a comment you must login or register.