Phospho-tau217 Springs Up in Synapses
Quick Links
Gotcha! Scientists spot where, and possibly how, p-tau217 first appears in the brain. In a postmortem study of individuals spanning the Alzheimer’s disease spectrum, researchers led by Michiko Sekiya and Koichi Iijima at the National Center for Geriatrics and Gerontology in Obu, Japan, report that the phosphorylated isoform crops up in neuronal synapses surrounding amyloid plaques. Published September 2 in Cell Reports, the findings hint that the biomarker might reflect an early synaptic response to amyloid buildup.
- P-tau217 appears adjacent to plaques early in AD.
- Its overlaps with p-tau202/205 and p-tau231.
- In synapses of both excitatory and inhibitory neurons.
In recent years, p-tau217 has emerged as the most promising blood-based biomarker for detecting the earliest traces of amyloid in the brain, closely tracking with amyloid load seen by PET—even in people with no signs of cognitive decline (Palmqvist et al., 2020). Now, scientists are taking a closer look to determine where this signal is coming from at the cellular level.
First authors Yu Hirota, Yasufumi Sakakibara, and colleagues looked at postmortem frontal cortex tissue stored at the Tokyo Metropolitan Institute for Geriatrics. They focused on gray matter from the middle frontal gyrus of 34 people with varying levels of AD pathology. This brain region was selected because plaques accumulate there before tangles do, allowing the effects of amyloid to be studied separately. Hirota divided the samples into three groups. Tissue from cognitively normal individuals who had had no plaques or tangles on autopsy; an early AD group, which they called “preclinical,” which included sample from people who were cognitively normal or mildly impaired when they died, had some amyloid plaques, but little or no tangle pathology; and an AD group consisted of individuals with clinical dementia and brains full of plaques and tangles.
Control brains showed no sign of p-tau217. In those with early amyloid pathology, however, a polyclonal anti-p-tau217 antibody from ThermoFisher, picked up specks of the isoform near plaques. These p-tau217 spots clustered around diffuse and dense-core deposits (image below), and their abundance climbed in step with plaque burden. They saw the same speckled pattern of staining with a separate antibody from Abcam. They didn’t test antibodies used in blood tests for p-tau217. In people who had AD, p-tau217 had spread into neuropil threads and tangles.
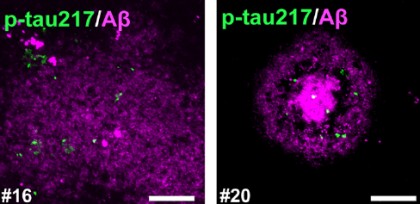
Phospho-Tau Embers. Phospho-tau217 (green) swirls around diffuse (left) and dense-core (right) plaques in the frontal cortex. [Image courtesy of Hirota et al., Cell Reports, 2025.]
Is p-tau217 alone, or do other markers tag along? To find out, the scientists co-stained tissue with the commonly used AT8 antibody, which recognizes p-tau202 and p-tau205. In brains with early-stage plaque and tangle pathology, both antibodies revealed flecks of p-tau surrounding plaques (image below). Quantification from three brains showed a hefty degree of overlap: on average, 70 percent of p-tau217 spots colocalized with AT8. Hirota and colleagues also found that p-tau231 gets in on the act, with 80 percent of p-tau217 spots sharing some p-tau231. In brains with later-stage pathology, all three markers showed up together in tangles.
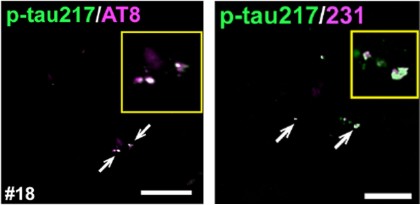
Partners in crime. During early stages of AD pathology p-tau217 (green) and p-tau202/205 (pink) co-localize (white arrows) in frontal cortex (left), as does p-tau217 and p-tau231 (right). [Image courtesy of Hirota et al., Cell Reports, 2025.]
Tau isoforms been shown to accumulate in the synapses of AD-model mice, prompting the scientists to ask whether phospho-tau might also be sitting in synapses in postmortem samples (Mondragón-Rodríguez et al., 2012; Ittner et al., 2010). Evidence of phospho-tau epitopes in synapses was also captured by electron microscopy in aging macaque monkeys, which, like humans, can develop AD-like pathology in old age (Datta et al., 2024; comment below). Sure enough, when Hirota stained frontal cortex sections from 10 early-stage AD brains, 38 percent of p-tau217 overlapped with the presynaptic marker synaptophysin, and another 31 percent colocalized with the postsynaptic marker PSD95 (image below). This did not depend on neuron type. P-tau217 overlapped with both excitatory and inhibitory presynaptic markers.
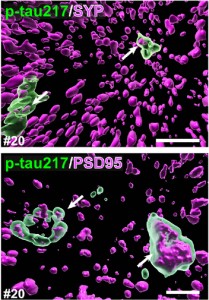
Location identified. In 3D confocal images, p-tau217 (green) cozies up to pre- (top) and post-synaptic (bottom) markers(purple). [Image courtesy of Hirota et al., Cell Reports, 2025.]
What is it doing there? “We do not consider p-tau217 in synapses to indicate damage,” Iijima told Alzforum. In support of that, they found that glia did not preferentially engulf p-tau217 containing synapses. “Rather, we see it as part of a response involving tau kinase activation.” On that note, Hirota also found that tau kinases, particularly active GSK3β, but also JNK and Cdk5, were enriched at p-tau217–positive synapses near plaques.
Sekiya noted that while p-tau217 is an excellent biomarker, the mechanisms by which it is released into the blood stream, need to be determined.
Hirota and colleagues also tracked the localization of p-tau181 in these brains. Unlike p-tau217, p-tau181 stained myelinated axons of glutamatergic and GABAergic neurons in the frontal cortex of control, early AD, and AD brains alike, with only a trend toward increased staining intensity as pathology worsened.
These results didn’t come as a surprise to the authors. “We expected these findings based on our previous work on mouse models,” Sekiya told Alzforum. They found that in mice, as in people, p-tau181 abounds in axons (Hirota, 2022). However, Stefan Kaeser, University of Tübingen, Germany, was concerned about the specificity of one of the antibodies used for p-tau181, namely AT270 (comment below). Scientists reported last year that it also binds neurofilament (Ellis et al., 2024).
For Fernando Gonzalez, University of Gothenburg, Sweden, the p-tau181 findings seemed reasonable. “We consistently observe that p-tau181 is more abundant than p-tau217,” he wrote to Alzforum (comment below). “[...] unlike at T181, phosphorylation at T217 is less physiological and more pathological in adults.” Likewise, Iijima believes p-tau217 is a more specific indicator of amyloid pathology in preclinical AD. “P-tau217 was rarely observed in healthy brains and only emerged after the formation of amyloid plaques,” he said.
Head to head, p-tau217 outperforms p-tau181 as a plasma marker—having higher sensitivity and specificity for AD (Palmqvist et al., 2020).—George R. Heaton.
George Heaton is a freelance writer in Durham, North Carolina.
References
Paper Citations
- Palmqvist S, Janelidze S, Quiroz YT, Zetterberg H, Lopera F, Stomrud E, Su Y, Chen Y, Serrano GE, Leuzy A, Mattsson-Carlgren N, Strandberg O, Smith R, Villegas A, Sepulveda-Falla D, Chai X, Proctor NK, Beach TG, Blennow K, Dage JL, Reiman EM, Hansson O. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020 Aug 25;324(8):772-781. PubMed.
- Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012 Sep 14;287(38):32040-53. Epub 2012 Jul 25 PubMed.
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22 PubMed.
- Datta D, Perone I, Wijegunawardana D, Liang F, Morozov YM, Arellano J, Duque A, Xie Z, van Dyck CH, Joyce MK, Arnsten AF. Nanoscale imaging of pT217-tau in aged rhesus macaque entorhinal and dorsolateral prefrontal cortex: Evidence of interneuronal trafficking and early-stage neurodegeneration. Alzheimers Dement. 2024 Apr;20(4):2843-2860. Epub 2024 Mar 6 PubMed.
- Hirota Y, Sakakibara Y, Ibaraki K, Takei K, Iijima KM, Sekiya M. Distinct brain pathologies associated with Alzheimer's disease biomarker-related phospho-tau 181 and phospho-tau 217 in App knock-in mouse models of amyloid-β amyloidosis. Brain Commun. 2022;4(6):fcac286. Epub 2022 Nov 6 PubMed.
- Ellis MJ, Lekka C, Holden KL, Tulmin H, Seedat F, O'Brien DP, Dhayal S, Zeissler ML, Knudsen JG, Kessler BM, Morgan NG, Todd JA, Richardson SJ, Stefana MI. Identification of high-performing antibodies for the reliable detection of Tau proteoforms by Western blotting and immunohistochemistry. Acta Neuropathol. 2024 May 18;147(1):87. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Hirota Y, Sakakibara Y, Morishima M, Sano T, Hara M, Arakawa A, Takao M, Murayama S, Saito Y, Sekiya M, Iijima KM. Biomarker-related phospho-tau217 appears in synapses around Aβ plaques prior to tau tangle in cerebral cortex of preclinical Alzheimer's disease. Cell Rep. 2025 Aug 26;:116203. Epub 2025 Aug 26 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
These findings are consistent with a previous publication showing that p-tau217 is associated with post-synaptic sites in cultured neurons, suggesting a role for T217 phosphorylation in synaptic impairment (Rajbanshi et al., 2023). However, from a practical perspective, the appearance of p-tau217 could be multifactorial and not limited solely to overproduction in AD as previously shown by multiple groups.
I think the extensive p-tau181 staining the authors see is reasonable. Using biofluid levels as a reference, we consistently observe that p-tau181 is more abundant than p-tau217. Although all p-tau forms seem to be physiologically produced, the lower abundance of fragments containing phosphorylated tau at T217 suggests that, unlike at T181, phosphorylation at T217 is less physiological and more pathological in adults.
Interestingly, this lower abundance of p-tau217 in both tissue and biofluids may explain why even small increases are more detectable when tau dysregulation occurs, as in AD and other conditions. Results from our lab using IP-MS show that p-tau217 is extremely sensitive for AD and even for secondary tau pathology, as in Creutzfeldt Jakob disease (Bentivenga et al., 2025; Lattanzio et al. 2017).
Additionally, CSF p-tau217 showed higher concordance with amyloid deposition. In the Norwegian Dementia Disease Initiation cohort, when using CSF p-tau217 as a tau status marker, we observed fewer amyloid-positive/tau-negative cases and more A+/T+ cases than when using p-tau181 as a tau measure. In contrast, there were more A−/T+ cases using p-tau181 (image below). I would say this aligns with the paper’s results, suggesting that in early AD pathology, p-tau217 increases almost simultaneously with amyloid, while p-tau181 significant increases appear later, possibly due to a higher baseline level.
[Image courtesy of Tormod Fladby and Bjorn-Eivind Kirsebom]
I find it interesting that the authors classify MCI as preclinical AD: “cognitively normal and mild cognitive impairment (MCI), Braak SP stage 1–2, NFT stage 0–3 (preclinical AD, Aβ+ /NFT−….” It is important to highlight that amyloid positivity does not always lead to clinical pathology. For example, in the DDI cohort, we have observed that cognitively normal A+ individuals (positive by CSF Aβ42/40 ratio) tend to remain cognitively unimpaired (Kirsebom et al., 2025).
The authors report that in preclinical AD brains, p-tau262 puncta appear around Aβ plaques, similar to p-tau217, and show strong colocalization with other tau epitopes (p-tau231 and p-tau181). They also highlight the importance of phosphorylation at Ser262 in promoting tau detachment from microtubules, initiating abnormal tau metabolism and further hyperphosphorylation. Our group is investigating the potential of CSF and plasma p-tau262 as a marker in AD.
Overall, despite the limitation of not having linked these neuropathological findings to CSF or plasma p-tau biomarker levels, the paper invites us to further explore the multiple aspects of AD pathology that p-tau217 captures. It would be particularly interesting to determine whether these findings are also reflected in biofluids and whether the p-tau217 associations track reliably with cognitive decline.
Yale Medical School
Hirota et al. found p-tau217, as well as other phosphorylated tau epitopes and activated kinases, at synapses in the prefrontal cortex (PFC) of patients with preclinical AD, especially within those with amyloid plaques. This is an important finding, as capturing early changes will be key to preventing AD. Elevations in plasma p-tau217 herald future disease, and indicate that there are large reservoirs of soluble p-tau217 accumulating as the disease process begins (Palmqvist 2020; Barthélemy 2024). However, this is usually hard to see in human brain, because soluble p-tau is not detected by tau-PET (Janelidze 2021), and it dephosphorylates rapidly (within 15min) postmortem, making it challenging to study in human brain (Wang 2015).
However, with perfusion fixation, we are able to capture p-tau217 and other tau epitopes, including p-tau181, p-tau231, and AT8 labelled tau, in aged macaques, who naturally develop amyloid and tau pathology with advanced age (Arnsten2021; Datta 2024). We then used high resolution immunoelectron microscopy (immunoEM) to see synaptic details with great clarity.
Our immunoEM validates what is reported here by Hirota et al., as we see p-tau217 at the synapse in aged macaque PFC. In particular, we see it in dendritic spines near the synapse and near calcium-storing smooth endoplasmic reticulum (SER) (image below), and occasionally in axon terminals. With high magnification, we can sometimes see it trafficking between neurons at the synapse, “seeding” a cortical network, and being exposed to the extracellular space where it can be captured in CSF and blood (image below) (Datta 2024). To date, we have only seen this at excitatory synapses, not at inhibitory synapses (Datta 2024; Paspalas 2018).
Hirota et al. hypothesize that the concentration of p-tau217 and other p-tau species near amyloid plaques may be due to Aβ42 driving tau hyperphosphorylation, and this likely involves Aβ42 increasing calcium actions near the synapse. For example, Aβ42 increases cytosolic calcium by creating calcium “pores” in the membrane, and by increasing internal calcium release from the SER (reviewed in Arnsten 2021; Arnsten 2025). With sufficient cytosolic calcium, there is activation of calpain-2, which cleaves and activates the main kinases that hyperphosphorylate tau, i.e. GSK3β and p25-cdk5 (Arnsten 2025), which Hirota et al. see in the preclinical AD synapses along with p-tau epitopes. Aggregated Aβ42 also inhibits phosphatase activity (Vintém 2009), which would sustain tau in a phosphorylated state. Thus, the mechanisms suggested by their data from human brains are consonant with a large literature from more basic studies.
However, there are also some important notes of caution. Hirota et al. show p-tau in parvalbumin-expressing axons in preclinical AD PFC, and these were interpreted as GABAergic axons. However, parvalbumin is also expressed in a large group of thalamo-cortical axons in primates (Jones 1998; Clascá 2012), which may be especially vulnerable to tau pathology (Braak 1991; Rüb 2002). Thus, one cannot assume that PV-expressing axons in human PFC are from local inhibitory GABA interneurons.
Hirota et al. focus much of their data on axons. It should be emphasized that although the old literature falsely states that tau is concentrated in axons rather than dendrites (Kanaan 2024), there is actually extensive tau in primate dendrites (Kanaan 2024), and indeed, tau pathology begins in dendrites and only reaches the axon after invading the soma (Braak 2018). It is noteworthy that axon terminals are often much larger than the small, thin spine heads that predominate in the primate PFC (Young 2014) (e.g. Fig. 1A), and with light microscopy, spines would be much harder to see than axon terminals. However, with the greater resolution of immunoEM in perfusion-fixed aged macaque PFC, we are able to see soluble p-tau217 concentrated in spines and dendrites rather than axons at early stages. There is extensive p-tau217 in dendrites, and it is likely toxic, interfering with endosomal trafficking and highly associated with autophagic degeneration6. This soluble p-tau217 in dendrites is likely lost in most human brain specimens due to extensive dephosphorylation postmortem (Wang 2015), including in the brains studied by Hirota and colleagues. However, we would speculate that the p-tau in synapses within amyloid plaques may be preserved from post-mortem dephosphorylation, where aggregated Aβ42 would inhibit phosphatase activity (Vintém 2009). Thus, like an ancient insect captured in amber, aggregated Aβ42 in plaques may preserve a process that allows us to see the earliest stages of tau pathology in human AD.
ImmunoEM of p-tau217 in synapses from aged macaque prefrontal cortex. A. ImmunoEM (left) showing p-tau217 (red arrowheads) in a dendritic spine (Sp, pseudocolored yellow), with p-tau217 concentrated on the calcium-storing SER in the spine (pseudocolored pink). The axon terminal (Ax, pseudocolored blue) contains a mitochondrion (Mit). The glutamate synapse is delineated by black arrows. B. Another example of p-tau217 in both a dendritic spine and in an axon terminal. The inset highlights the p-tau217 (red arrowheads) near the synapse, in an omega body (white arrowheads) consistent with trans-synaptic trafficking, “seeding” p-tau217 within a circuit. The scale bars indicate 200nm. From Datta et al, 2024.
University of Edinburgh
University of Ediburgh
This interesting study from Hirota and colleagues provides evidence in the human brain that p-tau217 staining is concentrated around plaques in Alzheimer’s disease and is associated with both synapses and distorted axons in the plaque microenvironment. Such findings are timely for the field, given the excitement around CSF and blood p-tau217 as biomarkers of early AD. Finding p-tau217 in synapses and dystrophic neurites around plaques provides hints as to why this biofluid marker of pathological tau signals that there is amyloid accumulation early in Alzheimer’s pathogenesis.
From our work and others in the field, we predict that synaptic tau is toxic to synapses and spreads through the brain by jumping through synapses (see, for example, Colom-Cadena et al., 2023). Both synapse death and extracellular tau moving through the synaptic cleft around plaques could be sources of extracellular tau that is detected in biofluids. Dystrophic neurites around plaques containing p-tau217 may also have damaged membranes that release tau into the interstitial fluid where it gets cleared into CSF and blood. However, without CSF/plasma data from the same tissue donors, against which this “brain data” can be correlated, such a conclusion cannot be confirmed with certainty, calling for further investigation.
References:
Colom-Cadena M, Davies C, Sirisi S, Lee JE, Simzer EM, Tzioras M, Querol-Vilaseca M, Sánchez-Aced É, Chang YY, Holt K, McGeachan RI, Rose J, Tulloch J, Wilkins L, Smith C, Andrian T, Belbin O, Pujals S, Horrocks MH, Lleó A, Spires-Jones TL. Synaptic oligomeric tau in Alzheimer's disease - A potential culprit in the spread of tau pathology through the brain. Neuron. 2023 Jul 19;111(14):2170-2183.e6. Epub 2023 May 15 PubMed.
Make a Comment
To make a comment you must login or register.