Protective ApoE Variant Quells Interferon Responses in Tauopathy
Quick Links
After the ApoE3-R136S “Christchurch” variant had held off the inevitable onslaught of Alzheimer’s disease by three decades in a woman with a pathogenic familial mutation, her brain was riddled with amyloid plaques but mostly free of neurofibrillary tangles. How did a point mutation in ApoE put a wrench in the AD cascade? According to a preprint posted April 28 on bioRxiv, it may calm an inflammatory wave. Scientists led by Li Gan, Weill Cornell Medical College in New York, report that in mice carrying the variant, microglia toned down their interferon responses, in part by not activating cGAS-STING, a cytosolic DNA sensor that can get tripped when nucleic acids leak from damaged organelles. This restraint spared synapses and prevented memory loss. Similarly, a cGAS inhibitor also protected synapses and downregulated the interferon response.
- In tauopathy mice, the ApoE3-R136S variant safeguards synapses and memory.
- It quells the microglial interferon response instigated by cGAS-STING pathway.
- A cGAS inhibitor similarly shielded synapses and squelched the interferon cascade.
Since the discovery that two copies of ApoE3-R136S (formally R154S) seemed to sever the tie between amyloid and tau pathologies, scientists have been digging for the mechanisms (Nov 2019 news; Sep 2022 conference news). Last year saw a report that the variant reduced seeding and toxicity of tau fibrils in mouse models of amyloidosis and tauopathy (Dec 2023 news).
For the current study, co-first authors Sarah Naguib, Eileen Torres, and Chloe Lopez-Lee and colleagues used CRISPR to replace both copies of the mouse ApoE gene with human ApoE3, or ApoE3-R136S. They then crossed these with PS19 mice, which express the FTD-causing P301S mutation in tau.
In tests of spatial learning and memory, 9-month-old male, but not female, ApoE3-PS19 mice were profoundly deficient compared to ApoE3 knock-ins on a wild-type background. The ApoE3-R136S staved off these memory problems, hence the scientists limited further analyses studies to males.
PS19 males expressing the Christchurch variant bore about half the tangle burden of their ApoE3 counterparts (image below). They also retained most of their synapses, while ApoE3-PS19 mice lost about a third. Despite these differences, microgliosis and astrogliosis unfolded similarly, regardless of which ApoE3 variant the mice expressed. Gan wondered if the type, rather than extent, of the glial response might help explain the variant’s protective effects.
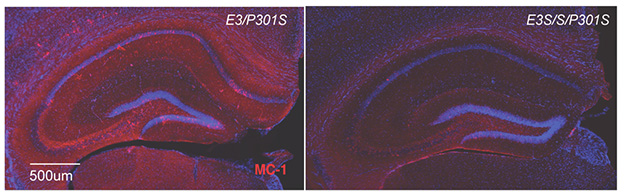
Tone Down Tau. ApoE3-PS19 mice (left) had twice as much aggregated tau (red) in the hippocampus as did ApoE3- R136S-PS19 mice (right). [Courtesy of Naguib et al., bioRxiv, 2024.]
To probe cell responses, the researchers surveyed the transcriptomes of single nuclei from the hippocampi of 9- to 10-month-old PS19 males. They detected effects of the variant on gene expression across multiple cell types. Notably, in R136S homozygotes, microglia downregulated genes involved in the type I interferon response. These included many genes upstream of the cascade, such as cyclic GMP-AMP synthase, aka cGAS. Previously, Gan had reported that this cytosolic DNA sensor became activated when tau fibrils damaged mitochondria, leaking mtDNA into the cytosol (Apr 2022 conference news). Once riled, cGAS triggers the protein STING to oligomerize, tripping off the interferon cascade (reviewed in Decout et al., 2021). Naguib and colleagues found dense clusters of the activated STING in ApoE3/PS19 mice, but hardly any in PS19 mice expressing the protective variant.
Tau fibrils triggered an inflammatory interferon cascade in primary microglia cells from ApoE3, but not in microglia from ApoE3-R136S mice. What’s more, R136S microglia rapidly degraded internalized tau. Gan told Alzforum that preliminary work in her lab suggests that a cGAS inhibitor similarly sped up digestion of tau fibrils within microglia, but doesn’t rule out that the variant might also encourage the cells to internalize tau. The original case study of the protective variant revealed that the R136S variant binds poorly to heparin sulfate proteoglycans, which could free up the surface receptors to latch onto tau (Arboleda-Velasquez et al., 2019).
Would inhibiting cGAS imitate the variant? To find out, the scientists fed 6-month-old ApoE3-PS19 mice chow laced with TDI6570, a brain-penetrant cGAS inhibitor. By 9 months of age, untreated ApoE3-PS19 mice had one-third fewer synapses than ApoE3 non-tauopathy mice or ApoE3-PS19 mice that had been treated with the cGAS inhibitor. Furthermore, single-nuclei transcriptomics revealed more overlap in the effects of the inhibitor and ApoE3-R136S in the mice across multiple cell types than would be expected by chance, Gan said. In microglia, several immune and interferon genes were among those commonly downregulated by both.
The findings suggest that turning down the type I interferon response explains part of the protection afforded by ApoE3-R136S, implying that some of the benefits of this rare mutation might be attained pharmacologically, with a cGAS inhibitor.
Gan believes that in tauopathies, mitochondrial DNA in microglia is the chief activator of cGAS. However, neurons express small amounts of the synthase and sustain DNA damage in tauopathies; they might also set it off. Her lab is generating conditional cGAS knockout mice to learn if other cells are involved, as well.
How ApoE3-R136S keeps the brakes on cGAS is unclear. The benefits could stem from bolstering microglial clean-up of tau fibrils, or from the way microglia respond to the pathology, Gan said.
Yun Chen and David Holtzman of Washington University in St. Louis have also hypothesized that ApoE3-R136S increases uptake and processing of tau fibrils. “What still eludes the field is the protection mechanism of ApoE-Christchurch at the cellular level,” they wrote (comment below). They think it is possible that the cGAS-STING-IFN pathway is involved, but believe reduced neurodegeneration might also contribute.
Yann Le Guen of Stanford University in Palo Alto begged off commenting on Gan’s study before it undergoes peer review, but noted that small numbers of mice and statistical techniques used in some of the snRNA-Seq experiments render the conclusions uncertain. He considers it unlikely that impaired binding to HSPGs explains why Christchurch protects, since he identified another HSPG-binding-deficient form of ApoE3, R145C (formally R163C), which either had no impact, or increased the risk of AD among African Americans, depending on their ApoE genotype (Mar 2023 news). Further studies are needed to understand how the protective variant stems the tide of tauopathy, Le Guen suggested.
Gan views the discovery of the protective variant as “nature’s gift.” “It is a highly resilient modifier, and if we can harness that, it may give us some way to reduce tau toxicity,” she said.—Jessica Shugart
References
Mutations Citations
News Citations
- Can an ApoE Mutation Halt Alzheimer’s Disease?
- In Brain With Christchurch Mutation, More ApoE3 Means Fewer Tangles
- APOE Christchurch Variant Tames Tangles and Gliosis in Mice
- Just Like Viruses, Tau Can Unleash Interferons
- In People with African Ancestry, ApoE3 Variant Ups Alzheimer's Risk
Paper Citations
- Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021 Sep;21(9):548-569. Epub 2021 Apr 8 PubMed.
- Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Naguib S, Torres ER, Lopez-Lee C, Fan L, Bhagwat M, Norman K, Lee SI, Zhu J, Ye P, Wong MY, Patel T, Mok SA, Luo W, Sinha S, Zhao M, Gong S, Gan L. APOE3-R136S mutation confers resilience against tau pathology via cGAS-STING-IFN inhibition. bioRxiv. 2024 Apr 28; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University School of Medicine
Washington University
In 2019, a case report (Arboleda-Velasquez et al., 2019) of a person who was homozygous for the APOE3 Christchurch (R136S) mutation caught the attention of many researchers studying APOE and AD, including our group. It might delay cognitive decline by 25 years, even in people with the autosomal-dominant Alzheimer’s disease mutation, PSEN1 E280A. The APOE3Ch homozygous carrier showed very limited tauopathy despite having massive amyloid deposition in the brain regions where this typically occurs in AD. Intrigued by this discrepancy between amyloid pathology and amyloid-induced tauopathy, we created a new humanized APOE3Ch mouse model and performed experiments on AD-tau seeding and spreading (Chen et al., 2024). We showed that APOE3Ch protects against amyloid-induced tauopathy. It appeared that the mechanism involves enhanced microglial responses toward neurotic plaque-tau aggregates. With in vitro experiments, we also demonstrated that a key molecular mechanism is the reduction of competitive receptor binding of APOE3Ch with LRP1/HSPG and consequently the enhancement of LRP1/HSPG-mediated tau uptake by myeloid cells. In addition, Nelson et al. introduced the Christchurch mutation into APOE4 (Nelson et al., 2023). In their study, they reported a decrease in both reactive microglia and astrocytes associated with protection against brain atrophy. Together, it is exciting that in different mouse models, R136S appears to protect amyloid-induced tauopathy and tau-induced neurodegeneration (in an APOE4 background), which prompts the idea to modify APOE to mimic the effect of the R136S variant as a therapeutic for AD.
One missing piece of the puzzle is a direct side-by-side comparison between APOE3 and APOE3Ch in a neurodegeneration model. Nelson et al. did not observe robust neuronal loss in their PS19 APOE3 mice compared with APOE4 mice (Nelson et al., 2023). In this new study, Naguib and Torres et al. compared PS19 APOE3Ch and PS19 APOE3 mice, showing a protection against P301S-mediated tauopathy and increased overall myelin in vivo with the APOECh variant. Interestingly, Naguib and Torres et al. observed a reduction of an IFN-I signature in microglia in the PS19 APOE3Ch mice. Furthermore, a cGAS inhibitor rescued synaptic loss in PS19 APOE3 mice restoring levels seen in thePS19 APOE3ch cohort. Consistent with the Gan lab’s previous publication on cGAS-STING-IFN pathway in tauopathy in vivo (Udeochu et al., 2023), this work further suggests that inhibitors targeting cGAS-STING pathway might be a promising pharmacological target for treating tauopathy and neurodegeneration.
However, what still eludes the field is the mechanism whereby protection of APOECh protects at the cellular level. At the biochemical level, there is no doubt that the APOE R136S variant reduces APOE receptor binding to LDLR, LRP1, and HSPG (Chen et al., 2024; Lalazar et al., 1988; Mah et al., 2023). At a cellular level, we hypothesized in our study, and showed that APOE3Ch impacts microglia responses toward amyloid and amyloid-induced tauopathy, likely due to the reduction in APOE3Ch’s receptor binding, because tau seeds and APOE lipoprotein particles compete for receptor binding. A recent study on the postmortem brain sample from the APOE3Ch homozygote carrier arrived at a similar conclusion, but focusing on LRP1 on astrocytes (Almeida et al., 2024). Nelson et al. suggested that the APOE3Ch protection rises from reduced uptake of paired helical fragments of tau in neurons. The molecular mechanism for this may be the same because the APOE variant difference was inhibited by heparin.
Gan and colleagues found that APOE3Ch microglia appeared to process aggregated tau fibrils more effectively, similar to what we observed in myeloid cells (Chen et al., 2024). While they previously showed that inhibition of the cGAS-STING-IFN pathway protects against tauopathy and tau-mediated neurodegeneration, it is not completely clear if APOE3Ch protects against neurodegeneration via the cGAS-STING-IFN pathway. First, reduction of microglial IFN signature in ApoECh mice may arise from the decreased neurodegeneration; second, while there are similarities there are also differences between cGAS-inhibitor- and APOE3Ch-driven changes in gene expression. This suggests that the same mechanism is not necessarily at play. Nonetheless, it is certainly possible that in the PS19 model APOE3Ch protects by influencing the cGAS-STING-IFN pathway.
Importantly, the protective effects of APOE3Ch in AD-related pathology has now been demonstrated in vivo by three independent research groups. It will be interesting to utilize this information to better understand the detailed mechanisms of its effects, as well as to test pharmacological therapies (Marino et al., 2024), gene therapies, or even cell therapies to investigate the translational potential of APOE3Ch-mediated protection against different aspects of AD pathology.
References:
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Chen Y, Song S, Parhizkar S, Lord J, Zhu Y, Strickland MR, Wang C, Park J, Tabor GT, Jiang H, Li K, Davis AA, Yuede CM, Colonna M, Ulrich JD, Holtzman DM. APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell. 2024 Jan 18;187(2):428-445.e20. Epub 2023 Dec 11 PubMed.
Nelson MR, Liu P, Agrawal A, Yip O, Blumenfeld J, Traglia M, Kim MJ, Koutsodendris N, Rao A, Grone B, Hao Y, Yoon SY, Xu Q, De Leon S, Choenyi T, Thomas R, Lopera F, Quiroz YT, Arboleda-Velasquez JF, Reiman EM, Mahley RW, Huang Y. The APOE-R136S mutation protects against APOE4-driven Tau pathology, neurodegeneration and neuroinflammation. Nat Neurosci. 2023 Dec;26(12):2104-2121. Epub 2023 Nov 13 PubMed.
Udeochu JC, Amin S, Huang Y, Fan L, Torres ER, Carling GK, Liu B, McGurran H, Coronas-Samano G, Kauwe G, Mousa GA, Wong MY, Ye P, Nagiri RK, Lo I, Holtzman J, Corona C, Yarahmady A, Gill MT, Raju RM, Mok SA, Gong S, Luo W, Zhao M, Tracy TE, Ratan RR, Tsai LH, Sinha SC, Gan L. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. 2023 May;26(5):737-750. Epub 2023 Apr 24 PubMed.
Lalazar A, Weisgraber KH, Rall SC Jr, Giladi H, Innerarity TL, Levanon AZ, Boyles JK, Amit B, Gorecki M, Mahley RW. Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. J Biol Chem. 1988 Mar 15;263(8):3542-5. PubMed.
Mah D, Zhu Y, Su G, Zhao J, Canning A, Gibson J, Song X, Stancanelli E, Xu Y, Zhang F, Linhardt RJ, Liu J, Wang L, Wang C. Apolipoprotein E Recognizes Alzheimer's Disease Associated 3-O Sulfation of Heparan Sulfate. Angew Chem Int Ed Engl. 2023 Jun 5;62(23):e202212636. Epub 2023 Apr 28 PubMed.
Almeida MC, Eger SJ, He C, Audouard M, Nikitina A, Glasauer SM, Han D, Mejía-Cupajita B, Acosta-Uribe J, Villalba-Moreno ND, Littau JL, Elcheikhali M, Rivera EK, Carrettiero DC, Villegas-Lanau CA, Sepulveda-Falla D, Lopera F, Kosik KS. Single-nucleus RNA sequencing demonstrates an autosomal dominant Alzheimer's disease profile and possible mechanisms of disease protection. Neuron. 2024 Jun 5;112(11):1778-1794.e7. Epub 2024 Feb 27 PubMed.
Marino C, Perez-Corredor P, O'Hare M, Heuer A, Chmielewska N, Gordon H, Chandrahas AS, Gonzalez-Buendia L, Delgado-Tirado S, Doan TH, Vanderleest TE, Arevalo-Alquichire S, Obar RA, Ortiz-Cordero C, Villegas A, Sepulveda-Falla D, Kim LA, Lopera F, Mahley R, Huang Y, Quiroz YT, Arboleda-Velasquez JF. APOE Christchurch-mimetic therapeutic antibody reduces APOE-mediated toxicity and tau phosphorylation. Alzheimers Dement. 2024 Feb;20(2):819-836. Epub 2023 Oct 4 PubMed.
Make a Comment
To make a comment you must login or register.