Synaptic Depression: The Missing Link between Aβ and Tau?
Quick Links
That Aβ begets tau pathology is the central premise of the amyloid cascade hypothesis of Alzheimer’s disease. But how exactly does the begetting happen? A paper published August 31 in Cell Reports posits that a crisis of synaptic strength links the two pathologies. Researchers led by Alexander Jeans of the University of Oxford, U.K., report that in hippocampal slices, Aβ oligomers goad neurons into excessively spewing out the neurotransmitter glutamate. In response, post-synaptic neurons enter a state of synaptic long-term depression (LTD), which, in turn, instigates an AD-like hyperphosphorylation of tau. Though individual parts of this pathway have been reported previously, the new study links them together, providing a potential mechanism connecting the two pathological hallmarks of AD.
- In hippocampal neurons, Aβ oligomers rev up glutamate release.
- Excess glutamate deepens long-term depression in post-synaptic neurons.
- LTD exacerbates tau phosphorylation in regions affected in Alzheimer's.
“This elegantly conducted paper provides valuable mechanistic insights that further reinforce growing evidence of synergy between the two hallmark proteins in Alzheimer’s disease, Aβ and tau,” wrote Samuel Harris and Marc Busche, of University College London, to Alzforum (comment below). “Notably, the authors present a novel mechanism that is potentially amenable to therapeutic targeting at very early stages of AD, before the tipping point at which tau pathology becomes established and, as a result of synergistic effects with Aβ, its effects prevail.”
A plethora of studies published over the past two decades have described how Aβ affects synaptic function. In 2003, researchers led by Roberto Malinow of University of California, San Diego, reported that synaptic activity boosts production of Aβ, which depresses synaptic transmission via NMDA receptors (Mar 2003 news). Later, studies led by Dennis Selkoe of Brigham and Women’s Hospital, Boston, found that Aβ oligomers prevented the uptake of glutamate, leading to an excess of the neurotransmitter and a subsequent squelching of synaptic signaling (Jun 2009 news). Other work supported the idea that Aβ aggregates boosts glutamate levels—a phenomenon that first led to hyperexcitability and then shut down synapses (May 2012 news and Aug 2019 news).
Could Aβ's synaptic exploits somehow set off tau hyperphosphorylation? This is the question first author Henry Taylor and colleagues addressed in the current study. Their hypothesis is rooted in the finding that synaptic LTD causes the phosphorylation of tau (Regan et al., 2015). They reasoned that excessive LTD caused by Aβ oligomers might push tau hyperphosphorylation into overdrive.
First, the researchers investigated how Aβ oligomers influenced glutamate release within the well-known Schaffer collateral circuit of the hippocampus. In hippocampal slice cultures, they stimulated the axons of CA3 neurons with electrical pulses, then monitored their release of glutamate-loaded synaptic vesicles onto CA1 neurons. Using a dye that becomes infused into synaptic vesicles as they are endocytosed by the post-synaptic cell, the researchers found that treating the slices with a preparation of Aβ oligomers boosted the probability of glutamate release from the stimulated neurons.
What would this glutamate glut do to LTD? To find out, the researchers stimulated CA3 neurons with low frequency electrical pulses—a standard way to provoke LTD. As expected, the post-synaptic CA1 neurons squelched synaptic signaling, by 22 percent. When Aβ oligomers were present, the LTD got bigger, snuffing synaptic currents by 58 percent. Aβ oligomers had no effect on LTD when the researchers blocked excessive glutamate release with a calcium channel blocker. So far, the findings suggested that Aβ oligomers enhance LTD in post-synaptic neurons by triggering a flood of glutamate from pre-synaptic neurons.
Now, for the big question: Would this extreme LTD exacerbate tau hyperphosphorylation? Indeed, incubating hippocampal slice cultures with Aβ oligomers triggered a boost in hyperphosphorylated and misfolded forms of tau, as measured by western blot with the AT8 antibody. Oligomers also stoked phosphorylation at threonine-231, a residue known to become phosphorylated early in AD. Importantly, these effects all but vanished when the researchers blocked excessive glutamate release with an inhibitor, or when they blocked NMDA receptors, which are required to start LTD. Finally, Taylor and colleagues found that they could instigate similar levels of tau hyperphosphorylation by inducing LTD chemically, with the inhibitor TBOA. This inhibitor is thought to cause LTD by engaging NMDA receptors that lie just outside of the synapse. They came to similar conclusions by inducing LTD using an optogenetic model. In all, the findings suggested that conditions that enhance LTD, including a rise in glutamate triggered by Aβ oligomers, lead to the pathological phosphorylation of tau.
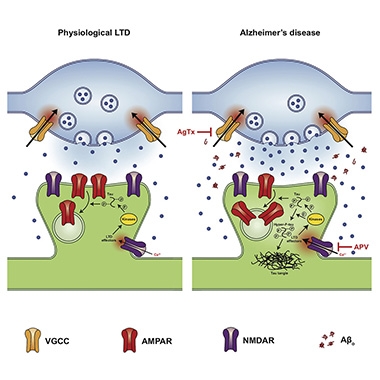
Depths of Depression. Under physiological conditions (left), glutamate (blue dots) stimulation of NMDA receptors triggers LTD, which leads to some phosphorylation of tau. With Aβ oligomers present (right), excess glutamate release exacerbates LTD, leading to hyperphosphorylation of tau. [Image courtesy of Taylor et al., Cell Reports, 2021.]
The findings illuminate the mechanisms by which Aβ's action at synapses may promote tau hyperphosphorylation, wrote Harris and Busche. Past studies have linked Aβ oligomers to tau phosphorylation via other mechanisms, including simulation of α2A-adrenergic receptors, or activation of the kinase Fyn within dendrites (Jan 2020 news and Jul 2017 conference news). Thickening the plot, Busche has reported that Aβ and tau aggregates exert opposing effects on synaptic transmission, implying a synaptic tug of war in the brain (Dec 2018 conference news and for review, Harris et al., 2020).
How might this Aβ-LTD-tau connection play out in the human brain? This remains an open question, Jeans told Alzforum. That said, the biology of the Schaffer collateral circuit is likely to apply to other regions of the brain, and Jean speculated that the relative vulnerability of different neurons to Aβ-induced LTD could dictate which cells develop tau pathology first.
Lindsay Welikovitch of Massachusetts General Hospital in Charlestown agreed that this cascade could be active in the AD brain. “It would be interesting to explore whether these effects are cell-autonomous, or whether neighboring glial cells, sensing changes in neurotransmission, may act as intermediaries,” she wrote. “In the context of previous works, this study also supports the possibility that induction of tau phosphorylation may initially serve to counter the effects of Aβ oligomers on neuronal activity, but becomes a ‘runaway train’ if chronically sustained.”
In support of the glia idea, a new human study analyzing networks of amyloid, tau, and activated microglia, all determined by PET, suggests that amyloid plaques prompt microglial activation, which in turn set tangle spread in motion (Sep 2021 news).—Jessica Shugart
References
News Citations
- Does Aβ Normally Rein in Excited Synapses?
- Neuronal Glutamate Fuels Aβ-induced LTD
- Soluble Aβ Takes Blame for Hyperactive Neurons in Mouse Brain
- Aβ Dimers Block Glutamate Uptake, Fire Up Synapses
- With a Shot of Adrenaline, Amyloid-β Sparks Tau Cascade
- Tau Silences, Aβ Inflames; Hitting Excitatory Synapses Hardest
- PET Firms Up Amyloid Cascade: Plaques, Inflammation, Tangles
Paper Citations
- Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, Ryu JH, Whitcomb DJ, Cho K. Tau Phosphorylation at Serine 396 Residue Is Required for Hippocampal LTD. J Neurosci. 2015 Mar 25;35(12):4804-12. PubMed.
- Harris SS, Wolf F, De Strooper B, Busche MA. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer's Disease. Neuron. 2020 Aug 5;107(3):417-435. Epub 2020 Jun 23 PubMed.
Other Citations
Further Reading
Primary Papers
- Taylor HB, Emptage NJ, Jeans AF. Long-term depression links amyloid-β to the pathological hyperphosphorylation of tau. Cell Rep. 2021 Aug 31;36(9):109638. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
University College London
This elegantly conducted study provides valuable mechanistic insights that reinforce growing evidence of synergy between the two hallmark proteins in Alzheimer’s disease, Aβ and tau (Busche and Hyman, 2020).
The authors present a novel mechanism that is potentially amenable to therapeutic targeting at very early stages of AD, before the tipping point at which tau pathology becomes established and, a result of synergistic effects with Aβ, its effects prevail (Harris et al., 2020; Busche et al., 2019).
The authors describe, for the first time, that Aβ oligomers in hippocampal slice cultures trigger an uptick in glutamate vesicular release. This increases the magnitude of long-term depression (LTD), which, in turn, instigates pathological tau hyperphosphorylation. These findings support previous reports that Aβ triggers tau pathology (e.g., Oddo et al., 2003; Choi et al., 2014) and provide further insights into the mechanisms by which the synaptic action of Aβ may promote tau hyperphosphorylation, e.g., via α2A-adrenergic receptors (Zhang et al., 2020), activation of Fyn (Li and Götz et al., 2017), and other effects, including blocking of glutamate reuptake, which also facilitates LTD (Li et al., 2009) and induction of neuronal hyperactivity in vivo (Busche et al., 2008; Harris et al., 2020). Crucially, these findings also point to a potential positive feedback loop of pathophysiology, in which Aβ-mediated increases in tau phosphorylation may recurrently promote Aβ toxicity (Ittner et al., 2010).
Perhaps most importantly, this research lends further support to the notion that targeting soluble, more pathogenic Aβ species is a valuable treatment strategy, and it raises the possibility that GSK-3β inhibition and NMDA receptor blockade may confer therapeutic benefits in early disease. For this to be confirmed, it will be necessary to validate these mechanistic insights in in vivo models of AD. Still, the current findings provide an important step forward in our understanding of the multifactorial and synergistic effects of Aβ and tau.
References:
Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer's disease. Nat Neurosci. 2020 Oct;23(10):1183-1193. Epub 2020 Aug 10 PubMed.
Harris SS, Wolf F, De Strooper B, Busche MA. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer's Disease. Neuron. 2020 Aug 5;107(3):417-435. Epub 2020 Jun 23 PubMed.
Busche MA, Wegmann S, Dujardin S, Commins C, Schiantarelli J, Klickstein N, Kamath TV, Carlson GA, Nelken I, Hyman BT. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci. 2019 Jan;22(1):57-64. Epub 2018 Dec 17 PubMed.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003 Jul 31;39(3):409-21. PubMed.
Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014 Nov 13;515(7526):274-8. Epub 2014 Oct 12 PubMed.
Zhang F, Gannon M, Chen Y, Yan S, Zhang S, Feng W, Tao J, Sha B, Liu Z, Saito T, Saido T, Keene CD, Jiao K, Roberson ED, Xu H, Wang Q. β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Sci Transl Med. 2020 Jan 15;12(526) PubMed.
Li C, Götz J. Somatodendritic accumulation of Tau in Alzheimer's disease is promoted by Fyn-mediated local protein translation. EMBO J. 2017 Nov 2;36(21):3120-3138. Epub 2017 Sep 1 PubMed.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009 Jun 25;62(6):788-801. PubMed.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
Harris SS, Wolf F, De Strooper B, Busche MA. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer's Disease. Neuron. 2020 Aug 5;107(3):417-435. Epub 2020 Jun 23 PubMed.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22 PubMed.
Massachusetts General Hospital and Harvard Medical School
This report outlines a clear pathway by which excessive Aβ oligomer-mediated glutamatergic signaling and resultant long-term depression may promote tau phosphorylation.
It is conceivable that the chronic effects of progressive amyloid accumulation on neurotransmission may similarly produce pathological changes in tau within the human brain. It would be interesting to explore whether these effects are cell-autonomous, or whether neighboring glial cells, sensing changes in neurotransmission, may act as intermediaries. In the context of previous works, this study also supports the possibility that induction of tau phosphorylation may initially serve to counter the effects of Aβ oligomer on neuronal activity, but becomes a “runaway train” if chronically sustained.
One is used to the fact that the abnormal hyperphosphorylation of tau is dealt with in a rather cursory fashion in the literature in spite of the fact that without it, tau pathology would not exist. Often it is simply ignored altogether. All too often, this gives license to superficial analysis, which is then squeezed into support of prefabricated concepts. This paper is such a case.
The claim of tau hyperphosphorylation rests solely on enhancement of AT8 reactivity, which by itself means nothing. The true authentic abnormal hyperphosphorylation of what is termed PHF-tau comes with a dramatic conformational change that is easily picked up on western blots. If properly done, a positive control reference is provided, as well.
None of that is seen here. The AT8 results could even be an artifact. As a result, the conclusions of an impact of electrophysiological activity on pathological tau phosphorylation, and certainly the claimed connection between the Aβ and tau pathologies, should be dismissed, as so many other claims before with equally sloppy analysis in vitro and in vivo.
There needs to be sounder craft in the field of tau hyperphosphorylation to stop the proliferation of confusion that has been going on for 30 years now.
University of Oxford
Reply to comment by Hanno Roder:
Unfortunately, the claims within our paper have been misinterpreted, or misunderstood. Dr. Roder describes specific misfolded conformations of tau, defined by altered migration on western blots, presumably under non-denaturing conditions. We are puzzled by the suggestion that this is the only meaningful definition of hyperphosphorylated tau, and the only useful way to study it, which is at odds with the majority of the tau literature.
Our paper investigates the role of synaptic activity on the phosphorylation of tau at a number of specific sites, carefully quantified on westerns with phosphorylation site-specific antibodies. Increasing levels of tau phosphorylation at these sites have been clearly associated with both tau misfolding (Bibow et al., 2011; Jeganathan et al., 2008) and, in a histopathological context, with the development and progression of AD (Braak et al., 1994; Mondragon-Rodriguez et al., 2014). Their pathological relevance is therefore clear.
Furthermore, the same sites have been shown not to be phosphorylated during normal, physiological synaptic activity of the type we are investigating (Regan et al., 2015). We certainly agree that extending our study by looking at effects on physical conformers of tau in addition would be interesting, and we would add, as other commenters have done above, that it will also be important to validate our findings in vivo.
However, this does not detract from the results as they stand, which address one of the major knowledge gaps in the field and set the stage for further study of this potentially important mechanism.
References:
Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. J Am Chem Soc. 2011 Oct 12;133(40):15842-5. PubMed.
Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87(6):554-67. PubMed.
Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008 Nov 14;283(46):32066-76. PubMed.
Mondragón-Rodríguez S, Perry G, Luna-Muñoz J, Acevedo-Aquino M, Williams S. Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer's disease and Down syndrome. Neuropathol Appl Neurobiol. 2013 Aug 23; PubMed.
Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, Ryu JH, Whitcomb DJ, Cho K. Tau Phosphorylation at Serine 396 Residue Is Required for Hippocampal LTD. J Neurosci. 2015 Mar 25;35(12):4804-12. PubMed.
Make a Comment
To make a comment you must login or register.