Synuclein Fibrils Trap Lysosome Repair Machinery
Quick Links
Scientists have uncovered a potential new mechanism whereby α-synuclein pathology spreads. Like black holes, large densities of α-synuclein pull in surrounding matter. Among the victims trapped in these aggregated masses are subunits of a protein conglomerate that goes by the mouthful “endosomal sorting complex required for transport-III.” ESCRT-III’s job is to patch damaged lysosomes. Now, Ulrich Hartl and colleagues at the Max Planck Institute of Biochemistry in Munich report that when synuclein captures ESCRT-III, lysosomal membranes become porous. Their contents, including α-synuclein fibrils, spill into the cytosol, potentially seeding new fibrils. The findings appeared on September 18 in Molecular Cell.
- ESCRT proteins repair lysosomal membranes.
- Fibrils of α-synuclein bind these proteins, exposing them to the proteasome.
- With repair compromised, lysosomes become porous, leaking synuclein fibrils into the cytosol.
“This is an elegant study, identifying mechanisms by which α-synuclein inclusions can lead to cellular toxicity ” said Michael Henderson, Van Andel Institute in Grand Rapids, Michigan. He was not involved in the research. “It highlights the importance of digging in the cellular “trash bin” for clues.”
One protein that consistently turns up in Lewy bodies is an ESCRT-III component called charged multivesicular body protein 2B (Tanikawa et al., 2012, Kurashige et al., 2012). CHMP2B is typically spread throughout the cytosol. To investigate how synuclein pathology affects this protein, first author Cole Sitron and colleagues expressed the highly aggregation-prone A53T α-synuclein mutant in HEK293 cells, then used the fat-soluble carrier lipofectamine to transfect them with fibrils to jump-start aggregation. Once that process started, CHMP2B was sucked into inclusions containing α-synuclein phosphorylated at serine-129, a marker of synuclein aggregates (image below). These inclusions also sequestered other ESCRT-III components, including CHMP2A, CHMP3, and CHMP4B.
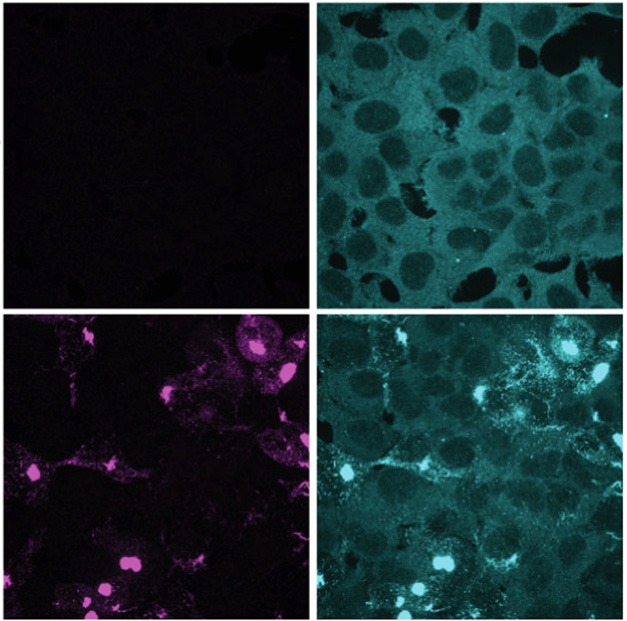
Synuclein Snares CHMP2B. Immunofluorescence images of HEK α-syn cells without (top) or with (bottom) fibril-induced aggregation. CHMP2B (cyan) is pulled into pS129-positive α-synuclein inclusions (purple). [Courtesy of Sitron et al., Molecular Cell, 2025.]
The scientists found that α-synuclein not only sequestered CHMP2B in inclusions, but lowered its overall levels in the cell, suggesting CHMP2B was degraded. Sitron suspected that ubiquitin ligases that target fibrils might catch CHMP2B in the crossfire, tagging it for degradation as well. Supporting this idea, blocking ubiquitination or proteosome activity restored CHMP2B levels.
“This is a new twist,” Wouter Peelaerts of KU Leuven in Belgium wrote to Alzforum (comment below). “Beyond the well-recognized sequestration of cellular proteins into Lewy bodies and neurites, fibrillar aggregates can actively deplete critical repair systems.”
Given the role of CHMP2B and other components of the ESCRT-III complex in lysosomal membrane repair, researchers asked whether loss of these proteins might weaken these organelles’ integrity. Using a Galectin-3 FRET reporter, which signals when sugars normally confined to lysosomes leak into the cytoplasm, they found that α-synuclein aggregation increased lysosomal damage. The authors attributed this effect to loss of ESCRT-III subunits, though they could not rule out that α-synuclein itself may directly damage the organelles.
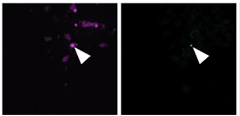
Fibril on the Loose. Immunofluorescence shows NeonGreen spots (green) co-localizing with pS129 α-synuclein (purple) as fibrils break into the cytoplasm. [Image courtesy of Sitron et al., Molecular Cell, 2025.]
What might loss of ESCRT-III mean for the spread of α-synuclein pathology? When fibrils enter cells, endosomes carry them to lysosomes for degradation. If those membranes are defective, the scientists reasoned, the fibrils could escape into the cytoplasm. To test this, they devised a split-NeonGreen assay. They fused one fragment of the fluorescent protein to α-synuclein, then assembled it into preformed fibrils. The complementary fragment was expressed in HEK293 cytoplasm. Fluorescence would flare only if fibrils breached lysosomes and reunited the two halves. The researchers added the tagged fibrils to either normal cells or cells preloaded with aggregates. Sure enough, fluorescence appeared only in the latter, where ESCRT-mediated membrane repair was impaired (image above).
The authors propose that when α-synuclein seeds reach the cytoplasm, they set off new waves of aggregation. Each fresh aggregate, in turn, could trap more ESCRT-III, making the cycle self-perpetuating (image below). “This finding provides a missing link between how α-synuclein first enters cells and how it spreads to amplify disease,” they write.
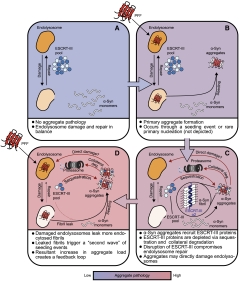
Vicious Cycle. Proposed model of how synuclein, by trapping ESCRT-III and compromising endolysosomal membrane repair, could promote its own pathology. [Image courtesy of Sitron et al., Molecular Cell, 2025.]
Researchers saw room for further investigation. “Structural information on how CHMP2B specifically binds fibrillar α-synuclein could provide a potential means to prevent this binding and ensuing endosome damage,” said Laura Volpicelli-Daley of the University of Alabama at Birmingham. She added that it will be interesting to see how genes linked to synucleinopathies, or environmental toxicants, influence α-synuclein–ESCRT-III interactions (comment below).
“As with all good studies, this one raises important new questions,” Henderson said. He highlighted several (comment below). For example, why do inclusion-bearing cells degrade CHMP2B? Are other sequestered proteins similarly lost? And why is CHMP2B localized to aggregates in the first place?
Besides tackling these questions, Sitron wants to identify other proteins that become collateral casualties of degradation, and to determine if and how this contributes to toxicity. The scientists also plan to explore why ESCRT-III proteins are especially prone to becoming trapped in aggregates—and not just of α-synuclein. The α-synuclein–binding region of ESCRT-III also latched onto Aβ42 fibrils, hinting that the motif may recognize amyloid structures more broadly. “We hope that this investigation will lead us to strategies that limit this vulnerability and break the cycle we described,” Sitron said.—George R. Heaton
George Heaton is a freelance writer in Durham, North Carolina.
References
Paper Citations
- Tanikawa S, Mori F, Tanji K, Kakita A, Takahashi H, Wakabayashi K. Endosomal sorting related protein CHMP2B is localized in Lewy bodies and glial cytoplasmic inclusions in α-synucleinopathy. Neurosci Lett. 2012 Oct 3;527(1):16-21. PubMed.
- Kurashige T, Takahashi T, Yamazaki Y, Hiji M, Izumi Y, Yamawaki T, Matsumoto M. Localization of CHMP2B-immunoreactivity in the brainstem of Lewy body disease. Neuropathology. 2012 Sep 19; PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Sitron CS, Trinkaus VA, Galesic A, Garhammer M, Yuste-Checa P, Dransfeld U, Feigenbutz D, Zhang J, Ivashko L, Dudanova I, Harper JW, Hartl FU. α-Synuclein aggregates inhibit ESCRT-III through sequestration and collateral degradation. Mol Cell. 2025 Sep 18;85(18):3505-3523.e17. Epub 2025 Sep 10 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Van Andel Institute
Lewy bodies and neurites rich in filamentous α-synuclein are hallmarks of Parkinson’s disease. While it is difficult to determine the kinetics of Lewy pathology toxicity in postmortem tissue, researchers have long searched for clues by examining other proteins localized to Lewy bodies. This intriguing study takes this approach with CHMP2B.
The authors start by demonstrating that α-synuclein inclusions induced by preformed fibrils in cell culture sequester CHMP2B. The team then leveraged an impressive set of cell biological tools to identify a specific binding between α-synuclein aggregates and CHMP2B. They showed that this interaction disrupts ESCRT assembly by causing CHMP2B degradation. Without proper ESCRT function, α-synuclein pathology increased.
This is an elegant study, identifying mechanisms by which α-synuclein inclusions can lead to cellular toxicity, and it highlights the importance of digging in the cellular “trash bin” for clues. As with all good studies, this one raises important new questions. Why is CHMP2B targeted for degradation in cells bearing inclusions? The authors suggest this could be collateral damage as the cell focuses on ubiquitinating α-synuclein. Are other sequestered proteins similarly lost? Why is CHMP2B localized to aggregates in the first place? Though a direct binding was observed in this study, it is unclear if this binding has a biological function. How does this play out over the years that neurons may bear aggregates instead of the days that these cells were challenged?
It is important to note that most experiments were performed in HEK cells due to the powerful tools available in those cells. However, α-synuclein fibrils require the addition of lipofectamine to bypass the endolysosomal system in HEK cells, missing a key biological pathway. Key findings were replicated in neurons, but there are certainly many future directions to pursue in neurons and in vivo.
KU Leuven
The study provides additional evidence for a cellular vulnerability in Parkinson’s disease: Seeded α-synuclein aggregation impairs the ESCRT machinery on which cells rely for membrane scission and repair. By dragging ESCRT proteins into fibrillar aggregates, α-synuclein appears to turn an essential repair system into collateral damage, potentially explaining aspects of Lewy pathology.
The authors used a clever lysosomal reporter assay to monitor ESCRT function under seeding conditions. They show that CHMP2B, an ESCRT component, binds α-synuclein fibrils via hydrophobic motifs in the C-terminus—and also found similar binding to Aβ. This reinforces the idea that CHMP2B recognizes amyloid folds by shape rather than sequence. Building on previous work showing that blocking the CHMP2B–α-synuclein interaction rescues pathology (Nim et al., 2023), the data make a case that this interface deserves attention for potential therapeutic development.
The consequences are interesting. Binding of α-synuclein fibrils to ESCRT does not merely sequester the machinery but also promotes collateral degradation. The cell may mistakenly target the ESCRT machinery together with the fibrils via ubiquitination. This is a new twist—beyond the well-recognized sequestration of cellular proteins into Lewy bodies and neurites, fibrillar aggregates can actively deplete critical repair systems. The result is impairment of endolysosomal repair, leakage across the membrane, and impaired degradation of fibrillar seeds. All three processes accelerate the escape of fibrils into the cytosol, where they can access monomeric α-synuclein to perpetuate seeded aggregation.
The work underscores that pathology requires active seeding of α-synuclein. The authors used a tunable α-synuclein expression system to induce pathology. Overexpression of monomeric α-synuclein alone had little effect, and preformed fibrils alone failed to cause ESCRT deficits. Only the combination, i.e., seeded aggregation, triggered dysfunction. Shutting off expression of α-synuclein relieved ESCRT defects, effectively rescuing cellular damage.
Although we need to keep in mind that these experiments were performed under saturating conditions, this finding is intriguing, as it suggests that lowering monomeric α-synuclein levels can reverse acute deficits and that reducing the monomeric pool of α-synuclein may be an actionable therapeutic strategy.
Together with the CHMP2B findings, the work highlights two complementary therapeutic angles: reducing the α-synuclein pool and blocking its harmful engagement with ESCRT. It will be interesting to see how these findings extend to more physiological systems, but both strategies hold promise for further exploration and intervention.
References:
Nim S, O'Hara DM, Corbi-Verge C, Perez-Riba A, Fujisawa K, Kapadia M, Chau H, Albanese F, Pawar G, De Snoo ML, Ngana SG, Kim J, El-Agnaf OM, Rennella E, Kay LE, Kalia SK, Kalia LV, Kim PM. Disrupting the α-synuclein-ESCRT interaction with a peptide inhibitor mitigates neurodegeneration in preclinical models of Parkinson's disease. Nat Commun. 2023 Apr 19;14(1):2150. PubMed.
University of Alabama- Birmingham
It has been known since the 1960s that Lewy pathology associates with degradative organelles (Duffy and Tennyson,1965; Forno and Norville, 1976; Watanabe et al., 1977). Our lab has shown that aggregates of α-synuclein selectively block axonal transport of endo-lysosomes and that signaling endosomes accumulate near these aggregates (Volpicelli-Daley et al., 2014). More recently, Lewy pathology composed of α-synuclein has been shown to co-localize with galectin-3, a marker of damage to endosomes and lysosomes. This overlap is not just a coincidence, as work from the labs of Edward Campbell and Dennis Dickson show the fibrils of α-synuclein internalize into neurons and damage endosomes and lysosomes (Freeman et al., 2013; Jiang et al., 2017). Neurons with damage lysosomes also show increased seeding of α-synuclein aggregates in the cytosol.
This current paper from Ulrich Hartl and colleagues demonstrates that the mechanism by which fibrils damage lysosomes depends on their interactions with the ESCRT-III complex, which mediates vesicle fission and membrane repair on endo-lysosomes. They show that α-synuclein fibrils, but not monomers, bind to the CHMP2B protein that is part of the ESCRT-III complex. Using HEK293 cells and iNeurons, they convincingly show that absence of CHMP2B, or mutations in CHMP2B associated with frontotemporal dementia, increase endolysosomal damage and enhance seeding of α-synuclein aggregation in the cytosol.
More structural information on how the CHMP2B a2 helix specifically binds fibrillar α-synuclein could provide a potential means to prevent this binding and ensuing endosome damage. It will also be interesting to see how genes implicated in Parkinson’s disease or dementia with Lewy bodies, or environmental toxins linked to Parkinson’s disease, can potentially influence α-synuclein/ESCRT-III binding.
References:
Duffy P, Tennyson V. Phase and Electron Microscopic Observations of Lewy Bodies and Melanin Granules in the Substantia Nigra and Locus Caeruleus in Parkinson's Disease. J Neuropathol Exp Neurol, July 1, 1965 Journal of Neuropathology and Experimental Neurology.
Forno LS, Norville RL. Ultrastructure of Lewy bodies in the stellate ganglion. Acta Neuropathol. 1976 Mar 30;34(3):183-97. PubMed.
Watanabe I, Vachal E, Tomita T. Dense core vesicles around the Lewy body in incidental Parkinson's disease: an electron microscopic study. Acta Neuropathol. 1977 Aug 16;39(2):173-5. PubMed.
Volpicelli-Daley LA, Gamble KL, Schultheiss CE, Riddle DM, West AB, Lee VM. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol Biol Cell. 2014 Dec 15;25(25):4010-23. Epub 2014 Oct 8 PubMed.
Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, Wiethoff CM, Campbell EM. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8(4):e62143. PubMed.
Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of α-synuclein aggregates. Sci Rep. 2017 Aug 9;7(1):7690. PubMed.
Roche
This study provides a compelling mechanistic explanation for how the process of α-synuclein aggregation, a key event in Parkinson's disease, impairs a vital cellular repair system, creating a destructive feedback loop that accelerates the disease process. The researchers demonstrate that fibrillar α-synuclein directly interacts with CHMP2B and other ESCRT-III proteins, sequesters them away from their site of function (endolysosomes), and as collateral damage, leads to degradation and depletion of functional ESCRT-III proteins.
This depletion initiates a vicious cycle. Without a functional ESCRT-III complex, the endolysosomes, which contain endocytosed α-synuclein fibrils, become increasingly damaged and leaky, allowing more fibrillar α-synuclein seeds to escape into the cytoplasm, where they trigger the aggregation of native α-synuclein. More aggregates lead to more sequestration and degradation of ESCRT-III, which in turn leads to more endolysosome damage and more seeding—a self-perpetuating feedback loop that amplifies the pathology.
Clarifying the mechanisms by which extracellular α-synuclein aggregates mediate and propagate its toxicity is particularly relevant. Interestingly, prasinezumab, an anti-α-synuclein antibody that targets extracellular α-synuclein, is positioned to be tested in a Phase 3 trial for early-stage Parkinson’s disease.
An interesting open question is whether boosting of residual ESCRT-III complex activity would limit or halt the toxic α-synuclein fibril mediated loop in its tracks. Currently, no tool compounds that boost ESCRT-III complex activity are available to test this hypothesis. Such compelling results, demonstrating the relevance of a functional lysosomal repair mechanism for healthy neurons, warrant efforts toward development of agents targeting lysosomal repair.
Make a Comment
To make a comment you must login or register.