TREM1 Muddles Myeloid Cell Metabolism and Memory in Old Mice
Quick Links
The macrophage and microglial receptor TREM1 whips up inflammation. In its tizzy, the receptor also perturbs myeloid cell metabolism and hastens cognitive decline in older wild-type mice and in two models of amyloidosis, according to scientists led by Katrin Andreasson at Stanford University, California. In a preprint uploaded to bioRxiv on March 11, they reported that knocking out TREM1 preserved glucose metabolism in peripheral macrophages and oxidative phosphorylation in neurons in the brain. Microglia from knockouts largely resisted Aβ oligomers, which caused wild-type microglia to shun oxidative phosphorylation for glycolysis. All told, TREM1 disrupts macrophage and microglial metabolism during normal aging and in AD, which seems to speed cognitive decline in both cases.
- TREM1 knockdown in 5xFAD mice protects neurons and preserves memory.
- Less than 5 percent of microglia express TREM1, implicating peripheral macrophages.
- They suppress glucose and nucleotide metabolism with age, but not if they lack TREM1.
“Linking TREM1 to microglial bioenergetics, and to myeloid cell metabolism in general, is exciting and opens a new avenue for potential therapeutic approaches in Alzheimer’s disease,” wrote Elizabeth Bradshaw and Kirstin Tamucci of Columbia University in New York (comment below). Indeed, another recent paper linked neuroinflammation in multiple sclerosis to a reversal of electron transport in microglial mitochondria (Mar 2024 news). Together, these studies suggest that inflammation and myeloid cell metabolic dysfunction may go hand in hand.
Because TREM1 amplifies inflammatory responses on myeloid cells, Andreasson and colleagues wondered if it might also play some role in Alzheimer’s disease. To find out, first author Edward Wilson and colleagues first asked if TREM1 affects cognitive decline that occurs during normal aging. He knocked out TREM1 in wild-type mice and tested their memories at 3 and 18 months. While the older mice expressing both TREM1 alleles had trouble recognizing novel objects and quickly escaping a Barnes maze, their counterparts lacking the receptor were as quick as the young mice. The knockout also spared mice from age-associated increases in cytokines and chemokines in the blood and brain.
What happened with myeloid cells in these knockouts? The scientists analyzed the transcriptomes of peripheral macrophages. Cells from old TREM1 knockout mice had almost the same profile as cells from young mice, with only 2.5 percent of their genes being differently expressed. Thirteen percent of genes in old wild-type macrophages were up/downregulated compared to cells from young mice. The results suggested knocking out TREM1 kept the macrophages youthful. Because less than 5 percent of mouse microglia express TREM1, the authors concluded that altered peripheral macrophage responses had protected the TREM1 knockouts from age-related cognitive decline.
Deleting TREM1 also kept metabolism in peripheral macrophages from old mice working like that in cells from young mice. The knockout suppressed glucose metabolism and improved mitochondrial respiration, boosting mitochondrial numbers and integrity.
“The deleterious role of basal TREM1 signaling in aging, driven by its disruption of myeloid glucose and nucleotide metabolism, is consistent with the general notion that systemic immune dysfunction is a major contributing factor to age-associated cognitive decline and neurodegeneration,” wrote Michal Schwartz of the Weizmann Institute of Science, Rehovot, Israel (comment below).
Might eliminating TREM1 protect against cognitive decline in AD? To test this, Wilson and colleagues turned to two mouse models of amyloidosis—5xFAD and APPSwe—knocking out TREM1 and testing memory at 6 or 18 months, respectively. Again, all knockouts, including 5xFAD TREM1 heterozygotes and APPSwe TREM1 hetero- and homozygotes, better recognized novel objects and escaped more quickly from a Barnes maze than did their counterparts with two TREM1 alleles.
Intriguingly, knocking out the gene had no effect on amyloid burden, though it did change how microglia respond to oligomeric Aβ42, at least in culture. Wild-type microglia suppressed oxidative phosphorylation and ramped up glycolysis, aka the Warburg effect, in the presence of the oligomers. However, in TREM1 knockout cells, it was metabolism as usual, with no letup in ATP production. The data suggested that TREM1-negative microglia might resist the effects of amyloid.
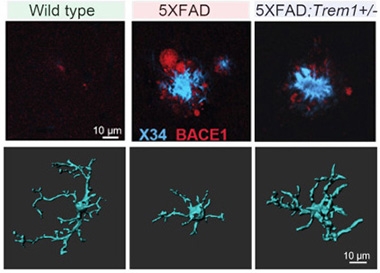
TREM1 Ramifications. Dystrophic neurites (red) ballooned around amyloid plaques (blue) in 5xFAD mice (top, middle), but less so in TREM1 knockouts (top, right), which retained ramified microglia (bottom). [Courtesy of Wilson et al., bioRxiv, 2024.]
Indeed, in knockout mice, microglia retained a mostly homeostatic, ramified form, extending cellular branches that were as long as those in wild-type mice, while microglia from 5xFAD controls blebbed up, a classic sign of activation (image at right). Curiously, transcriptomes of TREM1-deficient 5xFAD microglia were no different from control 5xFAD cells, both taking on a disease-associated microglia signature (Jun 2017 news). This made sense to Andreasson, since less than 5 percent of 5xFAD microglia expressed TREM1, just as in wild-type mice. Why, then, do the microglia retain their ramified shape? Andreasson thinks the peripheral immune cells are somehow involved, perhaps by communicating via molecules sent through the blood-brain barrier. Her lab is currently investigating how this might happen.
Neurons from 5xFAD TREM1 KOs seemed healthier, too, with neurites near cortical plaques being preserved. In mitochondria isolated from synaptosome fractions of APPSwe/TREM1 KOs, electrons skipped down the transport chain to drive ATP production as well as they did in organelles from wild-type mice. The mitochondria also seemed to metabolize glucose better, as its uptake in the hippocampus and thalamus was normalized, as measured by FDG-PET.
All told, the authors concluded that TREM1 triggers metabolic dysfunction in microglia and peripheral macrophages, the latter of which drives cognitive decline in aging and AD. In support of this, they found more TREM1 in the brains of people with AD than in healthy controls and that people who had more soluble TREM1 in their plasma were likelier to develop AD. “These findings suggest an alternative approach to diseases of aging, where myeloid cells might be reprogrammed to a healthier metabolic phenotype, providing a critical disease-modifying effect needed to slow or halt progression to AD,” they wrote.—Chelsea Weidman Burke
References
News Citations
- Reverse Electron Flow in Microglia Linked to Neuroinflammation
- Hot DAM: Specific Microglia Engulf Plaques
Research Models Citations
Further Reading
Primary Papers
- Andreasson KI, Wilson EN, Wang C, Xin M, Panchal M, Rabinowitz JD, Minhas PS, Swarovski MS, Benitez JA, Durairaj AS, Chaney A, Iweka CA, Buckwalter MS, Ennerfelt HE, Umans J, Huang J, Zera KA, McReynolds MR, Greicius MD, James ML, Mehta SS, LeGuen Y, Tan YJ, Zuckerman AJ, Blacher E, Gauba E, Serrano GE, Cropper H, Jain P, Liu Q. TREM1 disrupts myeloid bioenergetics and cognitive function in aging and Alzheimer disease models. 2024 Mar 11 10.1101/2024.03.05.583562 (version 1) bioRxiv.
Follow-On Reading
Papers
- Wilson EN, Wang C, Swarovski MS, Zera KA, Ennerfelt HE, Wang Q, Chaney A, Gauba E, Ramos Benitez JA, Le Guen Y, Minhas PS, Panchal M, Tan YJ, Blacher E, A Iweka C, Cropper H, Jain P, Liu Q, Mehta SS, Zuckerman AJ, Xin M, Umans J, Huang J, Durairaj AS, Serrano GE, Beach TG, Greicius MD, James ML, Buckwalter MS, McReynolds MR, Rabinowitz JD, Andreasson KI. TREM1 disrupts myeloid bioenergetics and cognitive function in aging and Alzheimer disease mouse models. Nat Neurosci. 2024 May;27(5):873-885. Epub 2024 Mar 27 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Columbia University
Columbia University
This paper highlights, and raises questions about, potential crosstalk between the peripheral immune system and central nervous system microglia and how this communication could be a powerful modulator of neurodegeneration. While the authors found very small numbers of TREM1+ myeloid cells in the mouse brain, concluding that peripheral TREM1 drives the phenotype, TREM1 KO microglia are resistant to bioenergetic changes caused by amyloid oligomer stimulation in vitro. It is important to note that once microglia are removed from their environment, they change their transcriptional profile to more resemble a non-specialized myeloid cell.
Linking TREM1 to microglial bioenergetics and to myeloid cell metabolism in general is very exciting. It opens a new avenue of potential therapeutic approaches for Alzheimer’s disease. The microglial bioenergetic experiments highlight the role these cells play in aging, and distinctly in diseases of aging, and emphasize these cells as major contributors to brain homeostasis. More work will be needed to determine what the contribution is of peripheral TREM1 and microglial TREM1 to the cognitive and behavioral phenotypes they observe in aged and APP mutant mice.
It was exciting to see a population of TREM1-positive microglia in the brains of these mice. Interestingly, this population grows in aged mice. Most surprising was the robust staining and immunoblotting of human TREM1 in the brains of both control and AD patients, suggesting the protein level is much higher than would be suspected by RNA-sequencing studies. Either the protein and RNA levels are not well correlated or indeed peripheral TREM1 is entering the brain and binding microglia specifically in aged and Alzheimer’s individuals. It was also exciting to see that their data suggest metabolic changes could be a driver of microglia phenotype and function. Intriguingly, they report increased brain glucose metabolism in the APPSwe model, while in their aging model and in human Alzheimer’s patients, there is a decrease in brain glucose metabolism. This speaks to both the difference between aging and diseases of aging, as well as what models should be used for studying neurodegeneration as it becomes clear microglial metabolism is a key component in diseases of aging.
At first glance, these results might appear to be counter to our findings that human monocytes decrease surface expression of TREM1 and have a reduced ratio of TREM1 to TREM2 on an AD pathology risk allele background (Chan et al., 2015). However, TREM1, like TREM2, exists as both a membrane-bound signaling molecule and a soluble receptor. Wilson et al. found that increased plasma levels of soluble TREM1 correlated with increased AD risk while the opposite was true for sTREM2. It is unclear if the sTREM1 comes from cleaved membrane-bound TREM1 or is a separate isoform, but there is potentially an inverse relationship between the soluble and membrane-bound forms. More work needs to be done to understand the function of sTREM1 compared to membrane-bound TREM1. Specifically, to determine if sTREM1 blocks membrane-bound TREM1 function as has been suggested (reviewed by Zhang et al., 2022).
References:
Chan G, White CC, Winn PA, Cimpean M, Replogle JM, Glick LR, Cuerdon NE, Ryan KJ, Johnson KA, Schneider JA, Bennett DA, Chibnik LB, Sperling RA, Bradshaw EM, De Jager PL. CD33 modulates TREM2: convergence of Alzheimer loci. Nat Neurosci. 2015 Nov;18(11):1556-8. Epub 2015 Sep 28 PubMed.
Zhang C, Kan X, Zhang B, Ni H, Shao J. The role of triggering receptor expressed on myeloid cells-1 (TREM-1) in central nervous system diseases. Mol Brain. 2022 Oct 22;15(1):84. PubMed.
Weizmann Institute of Science
The paper by Wilson et al. is extremely timely and important. It attributes a key role to myeloid cells in coping with aging in general, and age-related neurodegenerative diseases in particular, with an emphasis on Alzheimer’s disease, focusing on triggering receptor expressed on myeloid cells 1 (TREM1).
The role of myeloid cells, including blood-borne and resident microglia, in AD has been extensively explored, with a focus on TREM2. In this study, the authors attributed a novel negative role to TREM1 in cognitive decline in aging mice and in AD. Moreover, the effect of TREM1 on cognitive aging was found to be mediated predominantly by peripheral myeloid cells and not microglia.
Of note, TREM1 has been studied in cancer. Its expression is elevated in the tumor microenvironment, likely due to its expression by tumor-infiltrating myeloid cells. It is gratifying to see additional similarities between the cancer microenvironment and neurodegenerative disease.
The highly deleterious role of basal TREM1 signaling in aging, driven by its disruption of myeloid glucose and nucleotide metabolism, is consistent with the general notion that systemic immune dysfunction is a major contributing factor to age-associated cognitive decline and neurodegeneration. The present findings are in line with the notion that monocyte-derived macrophages are important players in defeating AD, and proposes that modifying circulating monocytes could provide a critical disease-modifying effect needed to slow or arrest progression to AD.
Make a Comment
To make a comment you must login or register.