Would ApoE Make a Better Therapeutic Target Than Aβ?
Quick Links
Although several anti-amyloid therapies in Alzheimer’s clinical trials melt away plaques, the process stresses blood vessels, causing tiny tears and fluid leakage into the brain. These morphological changes, dubbed amyloid-related imaging abnormalities, aka ARIA, can be detected by MRI. Could a therapy that goes after a different component of plaques clear them without causing ARIA?
- Anti-ApoE antibody HAE-4 clears plaque from a mouse model better than aducanumab did.
- Unlike aducanumab, HAE-4 causes no microhemorrhages.
- HAE-4 preferentially triggers microglia over astrocytes, and may be safer than Aβ immunotherapy.
In the February 17 Science Translational Medicine, researchers led by David Holtzman at Washington University, St. Louis, reported that HAE-4, an anti-ApoE antibody, mopped up plaques in a mouse model better than did the anti-amyloid antibody aducanumab. HAE-4 also cleared amyloid deposits from blood vessel walls, where aducanumab was unable to budge those, and it improved vascular function. Notably, HAE-4 did all this without causing microhemorrhages, hinting that this approach might be safer than using anti-amyloid antibodies. Some of these data were previously presented at the 2019 AD/PD conference (May 2019 conference news).
“Amyloid-bound ApoE appears to be a very good target for immunotherapy,” Holtzman told Alzforum. He has partnered with biotech company NextCure in Beltsville, Maryland, to develop the antibody for clinical use. Denali Therapeutics was initially working on HAE-4 but, according to Holtzman, made a business decision to pass on developing it clinically.
“This is an impressive and important paper,” said Gary Landreth of Indiana University School of Medicine in Indianapolis (full comment below). “[It] provides a clear rationale to pursue anti-ApoE based immunotherapy.” Guojun Bu at the Mayo Clinic in Jacksonville, Florida, agreed. Bu is optimistic about the potential of ApoE-directed therapies. “These data will spur new interest in targeting ApoE, and diversify our portfolio for treating AD,” Bu said.
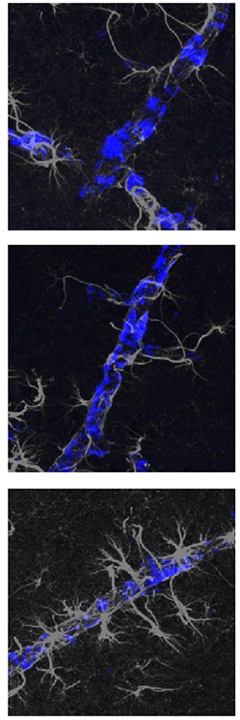
Astrocytes All Over Me. Brain blood vessels caked with amyloid (blue) attract few astrocytes (gray) in untreated AD mice (top), or in mice treated with an anti-ApoE antibody (middle), but many in aducanumab-treated mice (bottom). [Courtesy of Xiong et al., Science Translational Medicine/AAAS.]
The APOE4 allele is the strongest genetic risk factor for late-onset AD, and its protein binds Aβ fibrils and seeds plaques, particularly in blood vessels (Oct 2012 news; Dec 2017 news; Jun 2020 news). Its plaque-seeding capacity may be due to poor lipidation of the E4 variant protein, since boosting ApoE lipidation in mice suppresses plaque formation (Jun 2008 news; Sep 2014 news).
To exploit this difference, Holtzman’s team generated HAE-4 using a de-lipidated form of the human protein. In young AD mice expressing humanized ApoE4, treatment with HAE-4 curbed amyloid deposition without interfering with lipidated, physiological ApoE (Apr 2018 news).
The scientists compared HAE-4’s efficacy to known anti-amyloid treatments. First author Monica Xiong used a chimeric version of aducanumab that melds the antibody to a mouse Fc domain so it can interact with mouse microglia. She crossed 5XFAD mice with transgenic mice carrying homozygous human APOE4. Starting at 6 months of age, these animals develop parenchymal plaques as well as extensive deposits in blood vessels, known as cerebral amyloid angiopathy (CAA).
The researchers began treating these mice at 8 months of age, injecting 50 mg/kg of either HAE-4 or chimeric aducanumab into their bellies once per week for eight weeks. This works out to a human equivalent dose of about 4 mg/Kg, which is at the lower end of dosages used in clinical trials of aducanumab.
HAE-4 slashed plaque load in gray matter and in blood vessels in half. Even so, the treatment did not provoke microhemorrhages. For its part, aducanumab cut parenchymal plaque load by a quarter, which missed statistical significance, and barely budged CAA. Aducanumab-treated mice experienced larger and more frequent microhemorrhages than untreated controls.
Why the difference? The authors believed it might have something to do with the immune response. To test this, they injected older, i.e., 11-month-old mice with the antibodies four times over 10 days and examined acute responses. In mice receiving HAE-4, plaques in brain parenchyma and blood vessels attracted numerous activated, Iba1-positive microglia. These revved up disease-associated genes such as TREM2 and CST7, which are linked to plaque clearance (Jun 2017 news).
In mice receiving aducanumab, on the other hand, parenchymal and CAA plaques attracted fewer microglia, which failed to turn on disease genes. Instead, CAA plaques, in particular, triggered an astrocytic response, with numerous GFAP-positive astrocytes homing in on blood vessels (see image above). These astrocytes turned on inflammatory, complement-related proteins such as C3 and Serping1. Crucially, the amount of GFAP+ astrocytes around blood vessels correlated with microhemorrhages, implying they damaged the vessel. The aducanumab-treated mice also produced more pro-inflammatory cytokines, such as TNFα and IFNγ, in their bloodstreams compared to HAE-4-treated animals.
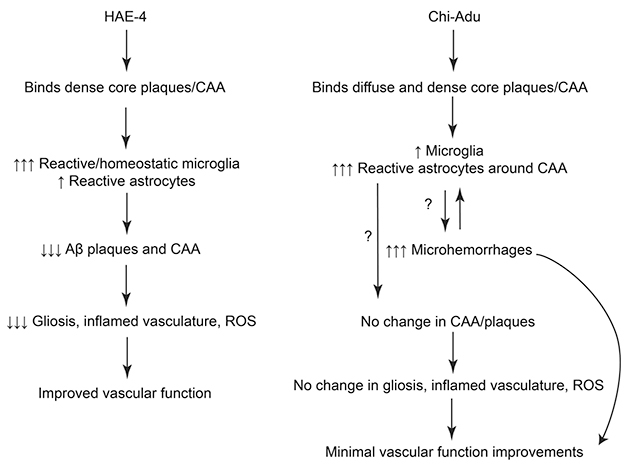
Different Paths. Anti-ApoE treatment (left) triggers a targeted immune response that removes fibrillar plaques and resolves quickly, while aducanumab (right) leads to a persistent inflammatory state. [Courtesy of Xiong et al., Science Translational Medicine/AAAS.]
The data suggest that aducanumab triggers a more generalized, damaging inflammation, whereas HAE-4 more narrowly targets plaque removal, Holtzman said. This may be because HAE-4 recognizes the poorly lipidated form of ApoE found in dense-core plaques and CAA. By pulling out this material, HAE-4 might help bust up plaques and clean out blood vessels. In contrast, aducanumab reacts with all forms of aggregated Aβ, including diffuse plaques, kindling a broader immune response. In addition, because aducanumab cannot clear CAA, solubilized Aβ does not drain as well from the brain. This backlogged material may create a sustained immune response, Holtzman speculated (see schematic above). Supporting this, inflammation stayed high over two months of aducanumab treatment, but fell back to baseline in HAE-4-treated animals.
Commenters agreed that astrocytes may damage blood vessels more than do microglia. “A lot of vascular inflammation relates to astrocyte activation,” Bu told Alzforum. Cristian Lasagna-Reeves at Indiana University, Indianapolis, has seen evidence for this as well. “In a mouse model for CAA, we recently demonstrated an increased activation of neurotoxic astrocytes associated to vascular amyloid deposits, but no major microglia reactivity,” he wrote (Taylor et al., 2020; full comment below).
HAE-4 treatment also strengthened the blood vessels’ tone. When the authors compared vessels with similar amyloid loads in the two treatment conditions, HAE-4 restored normal dilation and contraction, while aducanumab did not. This may be because de-lipidated ApoE damages blood vessels beyond its effect on amyloid, weakening the blood-brain barrier and causing leakage, Holtzman noted (Jan 2010 news; May 2012 news; May 2020 news). By removing this toxic ApoE, HAE-4 improved vascular function.
The next step will be to test whether HAE-4 treatment affects learning and memory, Holtzman said. It also remains to be seen what the antibody does in mouse models that develop tau pathology in addition to amyloid. ApoE4 worsens tau-related neurodegeneration as well, but in this condition, microglia contribute to the pathology (Apr 2017 news; Sep 2017 news; Oct 2019 news).
If anti-ApoE therapy were to pan out, it could have applications beyond AD, Holtzman believes. Some people develop CAA without parenchymal plaques. This is a debilitating condition that causes strokes, dementia, and early death. There is currently no treatment for it. “This approach could be a useful therapy for CAA, and would be worth testing,” Holtzman told Alzforum.—Madolyn Bowman Rogers
References
News Citations
- Antibodies Against Microglial Receptors TREM2 and CD33 Head to Trials
- ApoE4 Promotes Aβ Oligomerization
- ApoE4 Promotes Amyloidosis, But Only in Plaque-Free Mice
- Human Blood-Brain Barrier Model Blames Pericytes for CAA
- ApoE’s Secret Revealed? Protein Promotes Aβ Degradation
- Bexarotene’s Effects Vary by ApoE Genotype, Amyloid Pathology
- Human ApoE Antibody Nips Mouse Amyloid in the Bud
- Hot DAM: Specific Microglia Engulf Plaques
- Research Brief: Cerebral Blood Flow Ebbs In Aging E4 Carriers
- ApoE4 Makes Blood Vessels Leak, Could Kick Off Brain Damage
- Even Without Amyloid, ApoE4 Weakens Blood-Brain Barrier, Cognition
- ApoE and Tau: Unholy Alliance Spawns Neurodegeneration
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- In Tauopathy, ApoE Destroys Neurons Via Microglia
Research Models Citations
Paper Citations
- Taylor X, Cisternas P, You Y, You Y, Xiang S, Marambio Y, Zhang J, Vidal R, Lasagna-Reeves CA. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J Neuroinflammation. 2020 Jul 25;17(1):223. PubMed.
Further Reading
News
- More ApoE4 Means More Amyloid in Brains of Middle-Aged
- Lowering ApoE Brings Down Amyloid in Mice
- Upping Brain ApoE, Drug Treats Alzheimer's Mice
- San Francisco: Tweaking Brain ApoE Reduces Aβ, Symptoms
- Keystone: Therapies Around ApoE—Has Their Time Come?
- Has ApoE’s Time Come as a Therapeutic Target?
- Hats Off to ApoE—Key to Formation of Functional Amyloid
- ApoE Has Hand in Alzheimer’s Beyond Aβ, Beyond the Brain
- Without TREM2, Plaques Grow Fast in Mice, Have Less ApoE
Primary Papers
- Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, Gratuze M, Hoyle R, Bien-Ly N, Silverman AP, Sullivan PM, Watts RJ, Ulrich JD, Zipfel GJ, Holtzman DM. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci Transl Med. 2021 Feb 17;13(581) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Indiana University School of Medicine
This an impressive and important paper. Key to this study is the development of the HAE-4 anti-ApoE antibody and its specificity for poorly lipidated forms of ApoE, which are preferentially incorporated into amyloid plaques. The study is the quintessential product of the Holtzman lab, with expert experimental design, unambiguous outcomes, and clinically relevant conclusions.
There are a number of interesting findings. Notably, the chi-Adu is remarkably ineffective in this model in reducing plaque burden and CAA, while inducing microhemorrhages. Conversely, HAE-4 is effective at reduction of CAA as well as parenchymal plaques, with restoration of vascular function. The absence of an effect of chi-Adu on plaque load is different from its reported effect in clinical trials and prior animal studies, suggesting that the 5XE4 model may not faithfully model all aspects of the amyloid pathology, or that the actions of the chimeric antibody are distinct.
Overall, the paper provides a clear rationale to pursue anti-ApoE based immunotherapy.
Indiana University School of Medicine
This paper by Monica Xiong and colleagues elegantly demonstrates how anti-human ApoE immunotherapy efficiently reduced vascular and parenchymal amyloid deposits in the 5XFAD model expressing human ApoE4, whereas an anti-Aβ immunotherapy had no effect on cerebral amyloid angiopathy (CAA). Furthermore, the anti-Aβ antibody exacerbated microhemorrhage severity, whereas the anti-ApoE antibody didn’t stimulate microhemorrhages or rescue cerebrovascular dysfunction.
This is particularly relevant considering the disappointing outcomes of anti-Aβ immunotherapy clinical trials that resulted in ARIA, brain edema, and hemorrhages. Therefore, developing a therapeutic approach that safely removes amyloid accumulation not only from the parenchyma but also from the vasculature holds great promise for a possible treatment.
This study not only supports the development of anti-ApoE immunotherapy to safely reduce amyloid levels, but also opens the possibility of investigating antibody-based therapies targeting other known proteins closely associated with amyloid in CAA and AD.
Despite that, for the moment there isn’t a clear understanding of the mechanism by which this ApoE antibody safely removes CAA without stimulating microhemorrhages. This study underscores the importance of “the right amount” of inflammatory response associated with the treatment. It has to be strong enough to have an effect over amyloid load, but not strong enough to overstimulate an immune response detrimental to the vasculature. It’s necessary to determine this “right amount” of inflammatory response to further understand the dynamic immune response associated to AD.
On a similar point, future studies should focus not only on understanding the mechanisms involved in the immune response associated with amyloid plaques in AD but also with CAA. Interestingly, the authors showed how the anti-Aβ immunotherapy stimulated reactive astrocytes around CAA, which correlated with microhemorrhage severity, but very little reactive microglia, suggesting that proinflammatory cytokines known to stimulate astrogliosis may originate from another source different than microglia.
Interestingly, we recently demonstrated in a mouse model for CAA an increased activation of neurotoxic astrocytes associated to vascular amyloid deposits, but not major microglia reactivity (Taylor et al., 2020), suggesting as well that, in the context of CAA, proinflammatory cytokines known to stimulate astrogliosis may originate from another source. This supports the notion that the glia response associated to CAA-amyloid differs from the one associate to plaques in AD.
References:
Taylor X, Cisternas P, You Y, You Y, Xiang S, Marambio Y, Zhang J, Vidal R, Lasagna-Reeves CA. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J Neuroinflammation. 2020 Jul 25;17(1):223. PubMed.
Tel Aviv University
The Holtzman group's anti-ApoE antibody (HAE-4) recognizes human ApoE4 and ApoE3. This antibody binds specifically to non-lipidated human ApoE4 and ApoE3 and, when delivered to ApoE4-expressing mice, reduces Aβ deposition (Liao et al., 2018). These findings are now extended by the current paper, which shows that, unlike established anti-Aβ mAb, HAE- 4 also reduces cerebral amyloid angiopathy (CAA) and does not exacerbate microhemorrhages. These results suggest that targeting APOE in the core of both CAA and senile plaques could ameliorate amyloid pathology while protecting cerebrovascular integrity and function.
Since HAE-4 binds similarly to non-lipidated ApoE4 and ApoE3, it will be of great interest to assess the extent to which the HAE-4 driven immunotherapy is also apparent in non-ApoE4 carriers. This is particularly important as about half of AD patients are not ApoE4 carriers.
It is generally agreed that the pathological effects of ApoE4 are divided into Aβ-related and Aβ-independent mechanisms, and that ApoE4 is hypolipidated relative to the AD benign isoform ApoE3. Accordingly, it would be of interest to examine whether the ApoE4 epitope recognized by HAE-4 also plays a role in mediating non Aβ-related effects of ApoE4, such as downregulation of distinct receptors by ApoE4 in primary neuronal cultures.
References:
Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB, Robinson GO, Serrano JR, Silverman AP, Guo JL, Getz J, Henne K, Leyns CE, Gallardo G, Ulrich JD, Sullivan PM, Lerner EP, Hudry E, Sweeney ZK, Dennis MS, Hyman BT, Watts RJ, Holtzman DM. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest. 2018 May 1;128(5):2144-2155. Epub 2018 Mar 30 PubMed.
Make a Comment
To make a comment you must login or register.