Could CD33 Be the Microglial Target for Stimulating Phagocytosis?
Quick Links
Much of the research on microglia in Alzheimer’s disease has focused on the TREM2 receptor (see Part 4 of this series). But other microglial receptors play a hand in age- and injury-related activation as well (e.g., Apr 2019 news on CD22). One is CD33, a known AD gene that encodes another transmembrane receptor on these cells. There is a protective CD33 variant, and expression level and alternative splicing of the protein affect pathogenicity (e.g., Katsumata et al., 2019). Also known as Siglec-3, CD33 opposes TREM2 signaling and inhibits phagocytosis (Aug 2013 news; Griciuc et al., 2013).
- Suppressing CD33 stimulates phagocytosis, slows plaque growth.
- A protective CD33 mutation could be a gain of function that activates phagocytosis.
- The CD33 receptor acts as a dimer.
At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, Eloise Hudry of Massachusetts General Hospital, Boston, showcased experiments asking whether suppressing the receptor would unleash phagocytosis and thereby lessen pathology. She knocked down CD33 expression by a third in APP/PS1 mice by injecting an AAV-encoded microRNA targeting CD33 into the mice’s cerebral ventricles. In 8-month-old mice, which have well-established amyloidosis, CD33 reduction caused no change in plaques and only a modest drop in soluble Aβ42 three months later. In contrast, injections into 2-month-old mice slowed plaque growth, reducing total plaque area in the cortex and hippocampus by about one-quarter eight months later (see image below). Along with this, the researchers measured lower levels of some inflammatory markers, such as TNFα, IL1β, and TLR4. The results suggest microglia may have shifted from an inflammatory to a more phagocytic state, Hudry said.
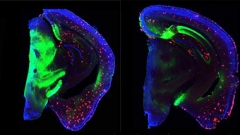
Curb CD33, Curb Plaque? Knocking down the microglial receptor (right) for eight months nudged amyloid plaque downward (red) compared with controls (left). Viral vector, green; nuclei, blue. [Courtesy of Eloise Hudry.]
Another presentation at AD/PD supported the idea that targeting CD33 might facilitate microglial phagocytosis. The protective variant of CD33 lacks a key effector domain that binds sialic acids and triggers intracellular signaling (Malik et al., 2013). Heyne (Cecilia) Lee of AbbVie in Ludwigshafen, Germany, used CRISPR to modify the CD33 gene in microglia made from human iPSCs. In some lines, the scientists cut out only this sialic acid-binding domain (D2-CD33), and in others they knocked out the entire gene. As expected, both cell lines phagocytosed more fluorescent zymosan particles (a yeast glycan) than wild-type microglia did. Intriguingly, while knockouts ate up twice as many particles as wild-types, D2-CD33 microglia did even better, consuming three times as many as wild-type. It is unclear why. A separate experiment lowering the amount of CD33 on the microglial surface also improved phagocytosis, underscoring the idea that targeting CD33 could be therapeutic (Zhao 2018).
However, a recent paper complicates the CD33 story. Researchers led by Steven Estus at the University of Kentucky, Lexington, examined a moderately rare, four-base-pair deletion that truncates the CD33 protein. This results in a complete loss of function in homozygotes and reduced expression in heterozygote carriers. In the IGAP database of 21,982 AD cases and 41,944 controls, the authors found that this deletion had no effect on AD risk. Estus told Alzforum that other unpublished genetic studies support this finding.
To Estus and colleagues, this implies that the D2-CD33 variant may protect not via loss of function, but rather via a gain of function in this receptor. Perhaps the shape of D2-CD33 allows it to bind intracellular activators rather than suppressors, in this way stimulating phagocytosis, Estus suggested. Specifically, the authors believe that instead of its canonical signaling through the SHP-1 adaptor, the mutant CD33 might bind Syk, allowing it to activate the same signaling pathway that TREM2 normally uses, thus enhancing phagocytosis (Estus et al., 2019). Intriguingly, this hypothesis dovetails with the in vitro findings from the AbbVie researchers.
In Lisbon, Peter St George-Hyslop of the University of Toronto, Canada, detailed how the risk and protective variants of CD33 interact. His group crystallized the extracellular domain of CD33 and determined its molecular structure. He found that CD33 forms dimers. The protective variant leads to alternative splicing, creating the form of CD33 that lacks the sialic acid binding site, while the normal variant produces a long protein containing this domain. Dimers of the long form are preferentially trafficking to the cell surface, explaining why these variants have a dominant effect, St George-Hyslop said. He believes the distinct shape of long-form dimers would allow them to bind large polysialylated ligands, and is currently trying to identify those ligands.
Some researchers are exploring the feasibility of targeting CD33. Luisa Quinti, working with Rudolph Tanzi at Massachusetts General Hospital, Boston, developed an assay using an immortalized mouse microglial cell line that expresses human CD33. She selected 37 drugs that previously had been found to affect microglia, and screened them for their ability to alter Aβ uptake and cytokine release in the cell line. Several drugs affected both; Quinti is now examining whether they act through CD33.
She also tested 23 antibodies against human CD33, and found seven that bound CD33 and dose-dependently suppressed its protein level. The assay may be a tool for finding CD33 modulators, Quinti concluded.—Madolyn Bowman Rogers
References
News Citations
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- CD22 Suppresses Microglial Phagocytosis—A New Therapeutic Target?
- Protective Microglial Gene Variant Promotes Phagocytosis
Paper Citations
- Katsumata Y, Nelson PT, Estus S, Alzheimer's Disease Neuroimaging Initiative (ADNI), Fardo DW. Translating Alzheimer's disease-associated polymorphisms into functional candidates: a survey of IGAP genes and SNPs. Neurobiol Aging. 2019 Feb;74:135-146. Epub 2018 Oct 23 PubMed.
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013 May 22;78(4):631-43. PubMed.
- Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, Estus S. CD33 Alzheimer's Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J Neurosci. 2013 Aug 14;33(33):13320-5. PubMed.
- Zhao L. CD33 in Alzheimer's Disease - Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology. 2018 Dec 12;:1-9. PubMed.
- Estus S, Shaw BC, Devanney N, Katsumata Y, Press EE, Fardo DW. Evaluation of CD33 as a genetic risk factor for Alzheimer's disease. Acta Neuropathol. 2019 Aug;138(2):187-199. Epub 2019 Apr 4 PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Sanders-Brown Center on Aging, University of Kentucky
The unpublished CD33 indel and AD genetics work mentioned in this news story were presented in October 2018 at the American Society for Human Genetics meeting by Adele Mitchell and colleagues. A link to their abstract is here.
Make a Comment
To make a comment you must login or register.