Do a Few “Master” Sites Control Tau Hyperphosphorylation?
Quick Links
Early in Alzheimer’s disease, phospho groups modify tau at multiple sites. What controls this process? In the July 6 Science Advances, researchers led by Arne Ittner at Flinders University, Adelaide, Australia, propose that some tau residues act as master regulators, with their phosphorylation facilitating the addition of phospho groups at multiple other serines and threonines.
- Phosphorylation at a handful of tau residues stimulates hyperphosphorylation at distant sites.
- In a mouse model of amyloidosis, blocking these “master” sites prevented tau hyperphosphorylation and preserved memory.
- Masking threonine 181 had the greatest effect, implicating p-tau181 as a key promoter of tau pathology.
These master sites—T50, T69, T111, T181, and T205—are concentrated in the N-terminal and proline-rich regions of the protein. Blocking phosphorylation at these residues in a mouse model of amyloidosis prevented tau hyperphosphorylation and maintained memory. The data suggest that targeting sites such as T181 could be a therapeutic strategy in AD, the authors noted.
Others were impressed by the work. “This is a comprehensive and remarkable investigation into the potential interdependency of different tau phosphorylation sites,” Amy Pooler at Sangamo Therapeutics, San Francisco, told Alzforum. Gerold Schmitt-Ulms at the University of Toronto agreed. “[This study] represents easily the most ambitious report of this phenomenon to date … I envision this work to become a benchmark for future studies,” he wrote.
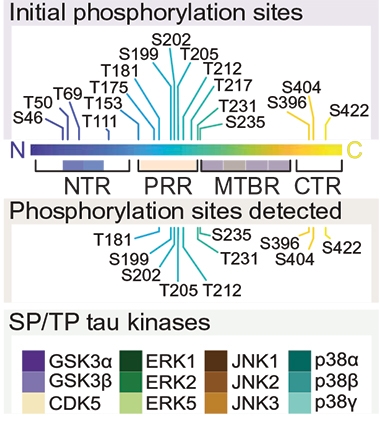
Interdependence Screen. Scientists mutated serine or threonine residues of tau (top) in HEK293 cells to mimic addition of a phospho group at 17 sites. After adding any of 12 different tau kinases (bottom), they measured effects on subsequent phosphorylation of 10 of the sites (middle). [Courtesy of Stefanoska et al., Science Advances/AAAS.]
Previous studies had found that tau phosphorylation could “prime” additional phosphorylation at nearby residues (Frame et al., 2001; Cho and Johnson, 2003). However, no one had done a systematic study of this phenomenon, nor looked for effects at more distal sites.
First author Kristie Stefanoska took this on. First she expressed human tau in HEK293 kidney cells in the presence of any of a dozen different tau kinases, including GSK3β, CDK5, and several isoforms of ERK, JNK, and MAP p38. Phosphorylations of tau occur most commonly at serine or threonine residues followed by prolines; there are 17 such sites. The authors mutated each of these sites in turn, changing serine to aspartate and threonine to glutamate to mimic addition of a phospho group, and then examined if those changes affected phosphorylation at the other sites. The complex dataset, with more than 2,000 combinations, identified five threonine residues as master phosphorylation sites: T50, T69, T111, T181, and T205. T181 was one of the most influential, promoting abundant phosphorylation at all 10 of the sites examined, including Alzheimer’s markers S199, S202, T205, and T231 (see image below). Phosphorylation at T217, another prominent AD marker, was not examined in the readout.
Notably, this relationship between different phosphorylation sites, which the authors dubbed interdependency, was most pronounced in the presence of the MAP kinase p38α, with other kinases more weakly promoting downstream phosphorylation. MAPK p38α is associated with Aβ-induced toxicity and is a therapeutic target in AD (e.g., see neflamapimod).
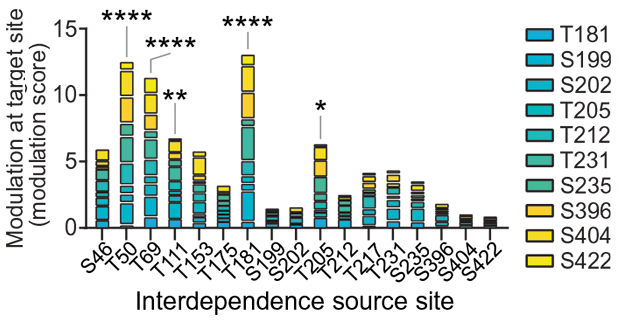
Hotspots. This plot shows how phosphorylations at any of 17 sites (x axis) affected addition of phospho groups at other sites represented by the vertical bars, which are color coded (right) to indicate site. The length of the bar indicates the amount of phosphorylation. Master sites T50, T69, and T181 and, to a lesser extent, T111 and T205, had the greatest effect. Asterisks indicate statistical significance. [Courtesy of Stefanoska et al., Science Advances/AAAS.]
Gail Johnson at the University of Rochester Medical Center, New York, said this screen represented a Herculean effort. “[It] yielded some new and very interesting findings … this long-range interdependence is very different from enhanced phosphorylation at a specific site due to a ‘priming’ event,” she wrote. Priming relies on close proximity between the phosphorylation sites, the idea being that if a kinase has already bound the protein, it will be likelier to phosphorylate again before it leaves. It remains unclear how phosphorylation might promote modification at distant sites. Nicolas Barthélemy at Washington University, St. Louis, speculated that modification in the N-terminal domain might decrease its interaction with the C-terminal region, perhaps allowing greater access for kinases.
Does this phosphorylation interdependency happen in vivo? In mice, replacing endogenous tau with a T205 phosphomimetic promoted phosphorylation of other sites, particularly T181, S396, and S422, suggesting this is a biological phenomenon. The authors chose T205 because it is an Alzheimer’s marker and binds the AT8 antibody that recognizes pathological tau.
Next, the authors wanted to know if blocking phosphorylation at master sites could protect mice. They replaced the threonines at positions 50, 69, or 181 with alanines, which cannot be phosphorylated. Then they used a viral vector to express each of these three constructs in APP23 mice that had the endogenous tau gene removed. All three suppressed hyperphosphorylation and preserved memory: Unlike APP23/tau-/- mice injected with the normal tau gene, mice harboring the tau mutants learned as well as did wild-types in the Morris water maze. Knocking out the p38a in neurons further suppressed tau hyperphosphorylation in mice expressing T69A or T181A, suggesting synergistic effects between the kinase and master regulatory sites.
Commenters said these findings need to be replicated, because expression from viral vectors can vary. The study raises new questions, such as the role tau phosphatases might play in interdependency and the temporal dynamics of the process, Pooler noted. Researchers also wondered if the dynamics of interdependency would be different between the six isoforms of tau; this study examined only the longest isoform, which includes two N-terminal inserts and four microtubule-binding repeat domains.
The findings may help explain why p-tau181 seems central in AD, Schmitt-Ulms said. Barthélemy suggested that because phosphorylation at T181 is much more common than at T50 or T69 in brain and body fluids, T181 is likely the most important master site. P-tau181 is an early AD biomarker, and plasma and cerebrospinal fluid levels have been linked to plaques, tangles, and disease progression (Mar 2020 news; Mar 2020 news; Jan 2021 news). For their part, the authors are exploring the therapeutic potential of a vaccine against p-tau181, which might remove it from mouse brain and reduce pathology.—Madolyn Bowman Rogers
References
Therapeutics Citations
Research Models Citations
News Citations
- Different CSF Phospho-Taus Match Distinct Changes in Brain Pathology
- A Phospho-Tau Plasma Assay for Alzheimer’s?
- Plasma P-Tau181 Predicts, Monitors Alzheimer’s Progression
Paper Citations
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001 Jun;7(6):1321-7. PubMed.
- Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003 Jan 3;278(1):187-93. PubMed.
Further Reading
News
- Is Tau Phosphorylation All Bad?
- BDNF Val66Met Hastens Tau Phosphorylation in Familial Alzheimer’s
- Inventory of Tau Modifications Hints at Undiscovered Functions
- Mounting Modifications Move Tau Toward Aggregation in Alzheimer’s Brain
- Different CSF Phospho-Taus Match Distinct Changes in Brain Pathology
- 217—The Best Phospho-Tau Marker for Alzheimer’s?
Primary Papers
- Stefanoska K, Gajwani M, Tan AR, Ahel HI, Asih PR, Volkerling A, Poljak A, Ittner A. Alzheimer's disease: Ablating single master site abolishes tau hyperphosphorylation. Sci Adv. 2022 Jul 8;8(27):eabl8809. Epub 2022 Jul 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Emory University
In this interesting study, the authors resolved the interdependent nature of tau phosphorylation sites and identified distinct residues as “master sites” that determine phosphorylation at multiple tau epitopes. Using a combination of genetic and biochemical approaches, they identified T50, T69, T181 and, to a lesser extent, T111 and T205 as master sites that, when phosphorylated, enhance distal tau phosphorylation.
This interdependence of tau phosphorylation can be directed by multiple tau kinases tested in the authors’ cellular assays. To this end, they show that p38 is one of the most central tau kinases linked to site-specific interdependence, which, when targeted significantly, reduced tau hyper-phosphorylation.
Ultimately this suggests that there is a hierarchy of tau phosphorylation that includes long-range effects between sites in distal regions of tau. Thus, by blocking a single master site, one could potentially block nearly all phosphorylation sites on tau including those most correlated to toxicity. Going forward, it will be interesting to see whether the master sites beyond T181, particularly those located on the N-terminal region of tau (T50 and T69), occur in early stages of human disease and, in turn, serve as peripheral biomarkers of pathology in CSF and/or plasma.
University of Toronto
This paper embodies the best in tau phosphorylation expertise and is a joy to read.
It is no secret that the activity of kinases toward specific phospho-acceptor sites can be influenced by other post-translational modifications, including phosphorylation. In the most basic form, a subset of kinases require a priming phosphorylation at a nearby site. The work by Stefanoska et al. transcends these basic observations by systematically studying the influence of tau phospho-mimetics on the phospho-occupancy of other tau phosphorylation sites. As such, the study is one of few that has shed light on the transition from tau phosphorylation to hyperphosphorylation. Because it limited the bulk of its systematic analyses to proline-directed tau phosphorylation sites in 293T cells, the study can only be considered a starting point; yet even so, it represents easily the most ambitious report of this phenomenon to date.
The data presented here advance our understanding of tau pathology, because the identification of master sites, including T181, promises to bring order to a contentious field of study. This may lead to a better understanding of why and how particular phosphorylation sites are more useful for diagnostics. I envision this work to become a benchmark for future studies aimed at understanding the relationship between a hierarchy and choreography of tau phospho-site occupancy and pathological tau folds in specific tauopathies.
In regard to potential therapeutic implications, the study makes a compelling case for the notion that kinases responsible for the phosphorylation of master sites, including p38α, should be given special consideration as therapeutic targets for the treatment of tauopathies.
University of Rochester
The paper by Stefanoska et al. is a very comprehensive analyses of how the phosphorylation of specific S/T-P sites in tau impact the phosphorylation of tau at other sites. Although the conceptual framework of interdependence, that is, the phosphorylation of one site impacting the phosphorylation at another, is not new, this study explores this idea in significantly more depth than previous studies. In their initial studies they used an HEK cell model where they expressed 17 different phosphomimetics (S/T→D/E) of tau and 12 different SP/TP kinases followed by SDS-PAGE and immunoblotting with 10 different phospho-tau antibodies for a total of 2,040 possible combinations—a Herculean effort! And it was an effort that yielded some new and very interesting findings. For example, they found that p38α was the kinase that was most strongly linked to interdependence. Indeed, NTR, as well as PRR, phosphomimetics significantly enhance tau phosphorylation by p38α, particularly in the CTR. As noted by the authors, this long-range interdependence is very different from enhanced phosphorylation at a specific site due to a “priming” event (e.g., phosphorylation of S235 results in increased phosphorylation of T231 by GSK3β). Interestingly, T205, which is in the AT8 epitope, was identified as a master site, as the T205A mutant resulted in phosphorylation at the majority of the phospho-epitopes examined being robustly decreased. In a 2020 publication (Ittner et al., 2020) they used CRISPR to make mice that expressed tauT205E/E and tauT205A/A. In this present study they used these mice to show that expression of tauT205E/E resulted in increased phosphorylation at several phosphoepitopes, while tauT205A/A resulted in decreased phosphorylation at some of the phospho sites. In the 2020 study they crossed these tauT205E/E and tauT205A/A CRISPRed mice with APP23 mice to study how phosphorylation at this site impacted disease progression. Upon analysis they found that lifespan in the APP23.tauT205E/E mice was significantly increased when compared with APP23.tauT205T/T (WT tau) and APP23.T205A/A mice. Further, learning and memory were significantly impaired in the APP23.tauT205A/A mice when compared with APP23.tauT205T/T mice, and significantly better in APP23.tauT205E/E mice. This finding is interesting in light of results of the present study in terms of T205 being one of the master sites for regulating phosphorylation at other sites. In contrast to the beneficial effect of T205 phosphorylation, phosphorylation of T181, another “master site,” seemed to have detrimental effects. In the present study they used AAVs to express wild-type tau (T181T) or T181A in the hippocampi of APP23.tau-/- mice and found that the presence of T181A tau resulted in significantly less impairment of memory compared to wild-type tau. T181 is also a master site, as expression of the T181A mutant also resulted in the reduction of the phosphorylation of several of the phospho-sites. A significant caveat to the comparison of the data with the T205 and T181 tau mutants is that the T205 mutants were CRISPRed in, while the T181 tau constructs were administered intrahippocampally using AAVs when the APP23.tau-/- mice were 2-4 months old. Nonetheless, these different results need to be considered when evaluating how phosphorylation of these “master sites” may impact not only the phosphorylation state of tau, but also its physiological and pathological function.
One last comment. In Figure 1A they present as part of their model a scenario where phosphorylation of one site reduces phosphorylation at other sites. Going forward actually identifying a site that when phosphorylated attenuates phosphorylation of other sites would be of significant interest.
References:
Ittner A, Asih PR, Tan AR, Prikas E, Bertz J, Stefanoska K, Lin Y, Volkerling AM, Ke YD, Delerue F, Ittner LM. Reduction of advanced tau-mediated memory deficits by the MAP kinase p38γ. Acta Neuropathol. 2020 Sep;140(3):279-294. Epub 2020 Jul 29 PubMed.
Sangamo Therapeutics
This is a comprehensive and remarkable investigation into the potential interdependency of different tau phosphorylation sites. With these data, new sets of questions pop up: What is the mechanism and, importantly, the role of tau phosphatases? What are the temporal dynamics of this process?
This paper primarily focused on the longest form of 4R tau, but are the dependencies altered in shorter tau isoforms? This work is exciting because understanding the interdependence of these phosphorylation sites may enable targeted therapies to disrupt development of pathologically phosphorylated forms of tau.
Washington University School of Medicine
This manuscript is really interesting. Ptau181 is one of the most abundant phospho-tau forms in brain and biofluids. We usually measure around 20 to 30 percent of phosphorylation on this site in normal tau (Barthelemy, 2019). Only S404 is more phosphorylated, at around 50 percent. Other sites found impactful by the authors (T50, T69, T111) are typically much less abundant, at around 1 percent or less. Together, tau sites’ phosphorylation abundance and these new results would indicate that ptau181 is likely the main modulator of other sites’ phosphorylation.
Obviously T181 becomes hyperphosphorylated in AD likely in response to amyloid plaques deposition. This hyperphosphorylation could help promote hyperphosphorylation on other sites. However, we have to keep in mind that ptau181 is not the earliest responder to plaque deposition. Sites such as T217, T231, and T111 might be impacted by hyperphosphorylation earlier.
The sites T50, T69 and T111 are found on tau domains regulated by exon2-3 alternative splicing. T111 is found on the three 0N 1N 2N isoforms, T50 and T69 in 1N and 2N. Our previous results suggest these sites might have different phosphorylation abundance depending on the isoforms considered. This could suggest other tau phosphorylated sites could be impacted by exon2-3 expression. The authors used the 2N isoforms for their investigation, which is also less abundant in brain. It would have been interesting to investigate if the results relative to T50, T69 and T111 are the same for 0N and 1N tau isoforms.
I am wondering if these results indicate that specific tau sites’ phosphorylation regulates, to some extent, the quite stochastic conformation of tau when it is associated with other proteins or structures, i.e., microtubules. A large proportion of the sites on tau’s N-terminal projection domain seems to impact other phosphorylation on the mid and C-terminal domains. I speculate that the Nter projection domain is interacting with these domains when not phosphorylated. When Nter is phosphorylated, this interaction would be lower, so Mid- and Cter domains would become more accessible and better substrates for kinases activity.
Though really comprehensive, the study would have benefitted from using some more quantitative approaches to measure tau phosphorylation. The relative abundance of each phosphorylated tau site compared to unphosphorylated (p-tau/t-tau) would have provided even more insights on ptau sites’ interconnection and the way they are impacted by the multiple kinases investigated in this study.
Also, site S208, which is an important component of the formation of the AT8- conformation specific epitope, was not screened. Some phosphorylated sites located on the MTBR domain, likely promoting tau disassembly from the microtubule, would have been interesting to investigate.
References:
Barthélemy NR, Mallipeddi N, Moiseyev P, Sato C, Bateman RJ. Tau Phosphorylation Rates Measured by Mass Spectrometry Differ in the Intracellular Brain vs. Extracellular Cerebrospinal Fluid Compartments and Are Differentially Affected by Alzheimer's Disease. Front Aging Neurosci. 2019;11:121. Epub 2019 May 21 PubMed.
Make a Comment
To make a comment you must login or register.