WANTED: Biomarkers for Drug Trials in Frontotemporal Dementia
Quick Links
At a recent workshop hosted for the Frontotemporal Dementia Study Group on March 31 and April 1 in Washington, D.C., researchers in academia, industry, and regulatory agencies universally agreed on one thing. They all want biomarkers to conduct clinical trials. Ideally, a whole bunch of them. Humbled by the hunt for Alzheimer’s drugs, scientists want FTD markers for several reasons.

For one, they need help picking the right patients for any particular trial. “It is critical to ascertain that patients in your trial have the disease you think they have. It was a big surprise to discover in the bapineuzumab trials that 30 percent of the patients did not have the disease the drug was designed to treat. Hopefully this will not be a problem in FTD,” said Geoffrey Kerchner of Genentech, South San Francisco. For another, they want pharmacokinetic and -dynamic markers to learn exactly what their drug does after they give it to people. “We must know if the compound was on board and hit its target. That is key. We must have that data so we can drop a target,” said Christoph Wiessner of Asceneuron. This biotech company in Lausanne, Switzerland, is hoping to start testing a small-molecule drug targeting a post-translational modification of tau in 2017.
From the Test Tube: Fluid Markers in FTD
Several candidate biomarkers exist, but how reliable are they? Kaj Blennow of the University of Gothenburg, Sweden, updated the group on the status of fluid markers in FTD. Cerebrospinal fluid markers for the molecular pathology of Alzheimer’s disease, i.e., Aβ42 and tau, have come a long way and are even approaching certification as clinical-grade diagnostic aids. Alas, researchers have known for at least a decade that neither total tau nor phospho-tau assays work in FTD (Hampel et al., 2004; Olsson et al., 2005). This was a head-scratcher—after all, tau pathology is the hallmark of many FTD disorders. Current tau assays are based on antibodies that recognize the protein’s mid-domain, and have been widely thought to measure “total” tau levels. However, recent mass spectrometry studies have shown that human CSF contains little full-length tau protein, but rather a range of N-terminal and mid-domain tau fragments. This raises the possibility that there are FTD-specific tau species in human CSF. If so, further mass spectrometry studies may identify fragments that could serve as the basis of FTD-specific tau assays, Blennow said (Portelius et al., 2008; Meredith et al., 2013).
A week after the FTSG meeting, on April 7, Blennow received Sweden’s 2016 Söderberg Prize in Medicine.
The new darling in the AD biomarkers field—neurogranin—also appears to be a non-starter in FTD. Neurogranin has long been known to be a component of dendritic spines, and new antibodies to it have led to tests that are rapidly gathering evidence supporting neurogranin as a CSF marker for synaptic degeneration in Alzheimer’s. Alas, the few studies that have thus far compared neurogranin across a range of dementing disorders suggest its signature increase may be specific to AD. This means the current AD fluid markers are useful indirectly, because they help exclude an FTD diagnosis, Blennow said.
Does anything work directly in FTD, then? Thus far, researchers are having more success with neurofilament light protein. NFL is a component of the cytoskeleton in large-caliber axons that course through white matter. Its concentration tends to rise in the CSF in instances of active neurodegeneration, including traumatic brain injury. NFL has been known since the year 2000 to go up in the CSF of people with FTD, but not AD, and it was rediscovered in 2014 by Adam Boxer’s group at University of California, San Francisco, and others. A comparison of 3,355 patients showed that CSF NFL increases about threefold in FTD, markedly higher than in other dementing illnesses, including AD, dementia with Lewy bodies, and vascular dementia (Sjogren et al., 2000; Scherling et al., 2014; Skillback, 2014).

The concentration of NFL in plasma amounts to less than a sugar cube dropped into an Olympic-size swimming pool. Could a blood test measure it accurately to spot FTD? [Courtesy of Kaj Blennow, U. Gothenburg.]
Could a blood test detect NFL? It’s a tall order. Compared to NFL’s concentration in the CSF (500 pg/ml), its concentration in plasma is but 10 pg/ml. This amounts to 3 percent of a sugar cube’s worth of NFL dissolved in an Olympic-size swimming pool that also contains 40 tons of other proteins, Blennow said. Abundant proteins such as CSF albumin would weigh in at 350 kg in the same pool.
But according to Blennow, a single molecule array (aka Simoa) assay can do it. The assay uses capture and biotin-tagged detection antibodies, as do ELISAs, but its bead-based technology and digital readout give it greater sensitivity. It measures NFL in serum down to a limit of 0.6 pg/ml, as compared to 78 pg/ml for ELISA and 16 pg/ml for Mesoscale. “You need these kinds of ultrasensitive technique to measure NFL in blood,” Blennow said.
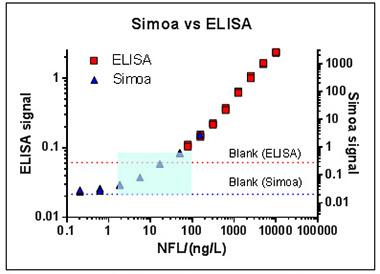
A single molecule array assay is more sensitive than ELISA, enabling scientists to detect neurofilament light chain in serum in the vanishingly low concentrations at which it occurs in human blood (blue box). [Courtesy of Kaj Blennow.]
Further data showed that NFL in blood correlates tightly with NFL in CSF; in other words, that serum NFL reflects brain pathology. With this reassurance, Blennow and collaborators at University College London tried the new test on blood samples of various FTD subgroups. Initial analyses of GENFI and other samples are beginning to show elevated serum NFL in all FTD subgroups, particularly in FTD-ALS and in progranulin mutation carriers, Blennow said. People with more severe symptoms tend to have higher serum NFL, and their brains shrink faster. This link between high serum NFL and fast decline is beginning to emerge in GENFI, and it has been published in the much larger ADNI cohort (Zetterberg et al., 2016). For clinical trials, NFL might therefore help researchers identify patients who are likely to progress fast. NFL in blood is also elevated in some parkinsonian disorders such as corticobasal syndrome (CBS), progressive supranuclear palsy (PSP), and multiple-system atrophy (MSA). Curiously, NFL shows no marked elevation in Parkinson’s, a slower-moving disease.
“Plasma NFL is a sensitive but unspecific progression marker for neurodegeneration,” Blennow said. As a candidate progression marker, could it flag a treatment effect in trials? There is no FTD data yet, but in multiple sclerosis, the answer is yes, at least according to a study in which blood NFL levels returned to normal in patients who responded to natalizumab antibody treatment (Gunnarsson et al., 2011). If blood NFL holds up in further testing, it may obviate the need for spinal taps in FTD, Blennow said.
What about blood proteomics and metabolomics? Most studies on multiple-ligand assays have started with up to 1,200 molecules and ended with panels of 18 or less that reportedly discriminate between groups. Typically, the changes in individual ligands are minute but collectively, their change can be significant in a statistical model. The small number of patients in these studies raises a concern of data over fitting, Blennow cautioned. Biomarker studies need to move beyond validating a panel by dividing the original cohort into a training and a validation set and adjusting the panel to optimize its discriminatory power. The way toward a robust assay is to take the panel to a totally different sample of people and try to validate the original finding. “Fix your panel and test it in a new cohort. If you can do that three, four times, then you have something,” Blennow said.
What about cytokines? Blennow noted that despite the growing interest in neuroinflammation in neurodegenerative disease research, general inflammation markers are difficult to measure. Cytokines are present at low levels of 1 to 2 pg/ml and tend to hover at the bottom of their assays’ calibration curves. Blennow recommended investigators run their samples alongside samples from multiple sclerosis or a chronic infectious disease such as Lyme to gain a sense of how cytokine changes in FTD compared to those in known neuroinflammatory conditions. “You typically see a slight change in AD or FTD compared to an enormous difference in MS,” Blennow said.
From the Scanner: MRI and PET
Bill Seeley of the University of California, San Francisco, compared structural and functional connectivity MRI. The former measures atrophy in particular brain areas. Its high signal-to-noise ratio and resolution make it attractive in FTD for making clinically defined subgroups more homogeneous, or for monitoring disease progression. Alas, the researchers also noted limitations of structural MRI as a potential outcome measure, including potential confounding fluid shifts in the brain. Fluid shifts have repeatedly produced puzzling results in some Alzheimer’s trials, where the brain temporarily shrank more in treated patients than in those on placebo (e.g., Jul 2004 conference news). Moreover, while researchers believe structural MRI might detect large effects of disease-modifying drugs, they do not expect an atrophy signal with symptomatic drugs.
All this is why high hopes have rested on imaging methods that measure brain function. Most recently, FTD researchers have focused on resting-state fMRI. In particular, a method called seed-based fMRI can illuminate the specific network of functionally connected brain areas that degenerate in a given FTD disorder, all of which are network diseases, said Seeley. For example, by using the dorsal midbrain tegmentum as a seed region of interest in healthy older people, scientists can see coordinated firing of a network that is a striking match to the atrophy pattern in PSP, and whose connectivity becomes degraded as PSP progresses, Seeley told the audience.
Alas, while elegant and promising, resting-state connectivity MRI is not ready for prime time, the scientists agreed. It is “noisier” than structural MRI and quite sensitive to head movement, a frequent problem when scanning people with a behavior disorder. Connectivity MRI is also sensitive to factors that subtly influence a person’s mental state, even simple things such as whether the patient had coffee prior to the scan.
Then again, older methods of imaging brain function, such as FDG PET, do pick up relatively rapid increases in brain function when a symptomatic drug works. Task-based fMRI tests are being evaluated in some trials. While none are robust and validated yet, new drugs such as AGB101 are targeting network excitability and will require a functional MRI readout. In D.C., investigators in both academia and industry emphasized that they need dynamic biomarkers that can tell them sooner than currently used readouts whether the drug at hand warrants a larger study or should be scrapped. Seeley noted that his research monitoring symptomatic FTD patients indicates structural MRI can flag decline over eight weeks. “We should develop these measures further,” agreed Brad Dickerson of Massachusetts General Hospital, Boston.
Summarizing PET imaging in FTD, Dickerson first noted that amyloid PET is already useful in dementia clinics to rule out Alzheimer’s disease. As an example, he told the case of a 62-year-old academic physician who one day tried to give grand rounds and sat down after 10 minutes, thinking he was finished. Initially he was thought to have AD, but his PiB scan came back negative, prompting more tests. His frontal lobe turned out to be hypometabolic on FDG PET and shrunken on structural MRI. He carried a progranulin mutation.
Tau PET is what everyone is is waiting for impatiently. With tau PET, researchers hope to see not only that patients in a trial of a tau-targeted drug have neurofibrillary tangles, but also whether the drug halts their spread. “For example, if you can show that you block progression of tau in PSP, we would be extremely excited as a company,” said Mark Forman of Merck in Philadelphia.
Alas, it’s early days. The best-studied tracer thus far, T807/AV1451, works well in Alzheimer’s but not FTD. “T807 is not the tracer we will use in FTD in the future, but it can teach us a lot along the way,” Dickerson said, to nods around the room. As Dickerson’s and other groups are scanning FTD patients they suspect of having tau pathology—for example symptomatic and presymptomatic tau mutation carriers, people with PSP—they are indeed seeing a T807 signal in expected areas of atrophy. The trouble is that they also see a signal in people whose FTD is likely due to TDP-43. More trouble: The T807 signal seen in living patients does not fully match results of autoradiography and other methods of postmortem tissue validation.
This may seem like a setback after the field had greeted tau PET with enthusiasm, but Dickerson said more research will surely clarify the binding properties of T807. What’s more, a handful of other tracers are nipping at its heels and will make for a good comparison. For one, the THK5351 tracer coming out of Tohoku University in Japan, looks promising, Dickerson said. Roche, Piramal, and Genentech all have started evaluating their tau tracers in people. Even Merck, a therapeutics giant that has no diagnostics business, has decided to make a tau PET tracer.
In D.C., Forman told the FTD Study Group that his company is developing the Merck tau PET tracer together with researchers at MGH in a joint project funded by the Alzheimer’s Drug Discovery Foundation. This tracer also has entered human trials, and first results are expected later this year. “It is new for us to develop an asset publicly and collaboratively. We do it because we want a high-quality tracer that will be available to the community and supports GENFI, ARTFL, and LEFFTDS. To us, these initiatives are moving the needle,” Forman said.
Besides tau, PET tracers are coming up for other targets, for example histone deacetylase (Strebl et al., 2015). While the neuroinflammation tracer PBR-28 is showing signals in neurodegeneration (see Zurcher et al., 2015), this ligand requires genotyping patients to understand their PET signal relative to control. The FTD Study Group agreed that the field needs to develop new imaging and fluid markers of neuroinflammation, ideally specific markers for well-characterized targets.
Over and over in discussion, industry, academic, and regulatory researchers called for rigor in biomarker development and for learning the bitter lessons of AD. They emphasized that any drug moving forward should have strong evidence of target engagement. “For this, we have to have quick response measures,” said Philipp von Rosenstiel of Biogen in Cambridge, Massachusetts.
Currently in Alzheimer’s disease, companies are running large, long Phase 3 trials to look for small signals. This is in part because there are no validated outcome biomarkers and in part because companies are reluctant to give up candidate drugs that already cost so much time and money to move through Phase 2. The FTD Study Group hopes that with better biomarkers, FTD trials will more efficiently tell researchers whether the investigational drug at hand works. This is especially critical in a set of rare spectrum disorders, where trials will have to stay small. “We have many examples in AD where we said we can power up and do a muscular trial and get over these problems. But that is wrong. We need to have biomarkers so we can interpret trial results,” said Howard Feldman of the University of California, San Diego, who took over the Alzheimer’s Disease Cooperative Study.
Most people with FTD are younger than late-onset AD patients, hence they have fewer age-related morbidities that muddy the disease picture and make them prone to side effects. But just like in AD, intervening early is important and, at least in sporadic cases, that is impossible without biomarkers. “We are all in line with the notion that by the time a patient comes to clinic and a simple interview makes clear they have FTD, it may be too late. Hopefully we can learn to diagnose patients when their clinical symptoms are very subtle. The field is clearly aligned around that goal,” Kerchner said.
All in all, the upshot of the day was that some FTD biomarkers are already useful in exploratory trials and should be embedded in all FTD trials to generate data, but as of 2016, none are close to receiving formal regulatory qualification for a particular context of use. To learn about the regulatory discussion, see Part 3 of this series.—Gabrielle Strobel
References
Therapeutics Citations
News Citations
- Philadelphia: Can a Shrinking Brain Be Good for You?
- Regulators Tell Frontotemporal Dementia Community: We Play on Your Team
Paper Citations
- Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, Sjoegren M, DeBernardis J, Kerkman D, Ishiguro K, Ohno H, Vanmechelen E, Vanderstichele H, McCulloch C, Moller HJ, Davies P, Blennow K. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004 Jan;61(1):95-102. PubMed.
- Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005 Feb;51(2):336-45. Epub 2004 Nov 24 PubMed.
- Portelius E, Hansson SF, Tran AJ, Zetterberg H, Grognet P, Vanmechelen E, Höglund K, Brinkmalm G, Westman-Brinkmalm A, Nordhoff E, Blennow K, Gobom J. Characterization of tau in cerebrospinal fluid using mass spectrometry. J Proteome Res. 2008 May;7(5):2114-20. PubMed.
- Meredith JE, Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ, Slemmon JR, Portelius E, Zetterberg H, Blennow K, Soares H, Ahlijanian M, Albright CF. Characterization of Novel CSF Tau and ptau Biomarkers for Alzheimer's Disease. PLoS One. 2013;8(10):e76523. PubMed.
- Sjögren M, Rosengren L, Minthon L, Davidsson P, Blennow K, Wallin A. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology. 2000 May 23;54(10):1960-4. PubMed.
- Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, Kramer JH, Rabinovici G, Ahlijanian M, Miller BL, Seeley W, Grinberg LT, Rosen H, Meredith J Jr, Boxer AL. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014 Jan;75(1):116-26. Epub 2014 Jan 2 PubMed.
- Skillbäck T, Farahmand B, Bartlett JW, Rosén C, Mattsson N, Nägga K, Kilander L, Religa D, Wimo A, Winblad B, Rosengren L, Schott JM, Blennow K, Eriksdotter M, Zetterberg H. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology. 2014 Nov 18;83(21):1945-53. Epub 2014 Oct 22 PubMed.
- Zetterberg H, Skillbäck T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM, Weiner MW, Blennow K, Alzheimer’s Disease Neuroimaging Initiative. Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol. 2016 Jan 1;73(1):60-7. PubMed.
- Gunnarsson M, Malmeström C, Axelsson M, Sundström P, Dahle C, Vrethem M, Olsson T, Piehl F, Norgren N, Rosengren L, Svenningsson A, Lycke J. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann Neurol. 2011 Jan;69(1):83-9. PubMed.
- Strebl MG, Wang C, Schroeder FA, Placzek MS, Wey HY, Van de Bittner GC, Neelamegam R, Hooker JM. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem Neurosci. 2015 Dec 21; PubMed.
- Zürcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, Akeju O, Catana C, Rosen BR, Cudkowicz ME, Hooker JM, Atassi N. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [(11)C]-PBR28. Neuroimage Clin. 2015;7:409-14. Epub 2015 Jan 19 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.