Overactive Kinase May Drive Aβ to Depress Synapses
Quick Links
Research suggests that Aβ reduces the excitability of synapses, causing long-term depression. How? In the May 10 Science Signaling, scientists led by Roberto Malinow and Alexandra Newton at the University of California, San Diego, and Rudolph Tanzi at Massachusetts General Hospital, Charlestown, report that protein kinase C α (PKCα) may be involved. In mice, blocking PKCα prevented the synaptic depression caused by Aβ; conversely, more-active variants of the kinase that occur in some families with late-onset Alzheimer’s disease may predispose to the disease.
Researchers had previously found that chronically activating PKC promotes Aβ production from the amyloid precursor protein. “This new synaptic role for PKC in modulating Aβ-induced synaptic depression potentially yields a genetic double whammy if the overactive PKC clinical mutation drives both Aβ generation and Aβ-induced synaptic depression,” noted Samuel Gandy, Icahn School of Medicine at Mount Sinai, New York. Others questioned the strength of the genetic data.
Open to change.
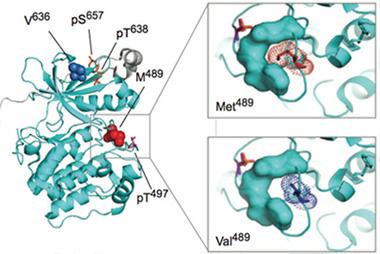
In PKCα (left), replacing methionine (red, top right) with valine (purple, bottom right) makes the active site more accessible. [Courtesy of Science Signaling/AAAS]
The PKC family phosphorylates proteins involved in cell proliferation and death, as well as in gene transcription and translation. These kinases have been implicated in the regulation of learning and memory. They have been studied for a role in AD, possibly via stimulation of APP synthesis and processing (da Cruz e Silva et al., 2009; Lane et al., 2010; Kim et al., 2011; Kinouchi et al., 1995). Most of this work was done in model systems, but recently, Japanese researchers found that PKC phosphorylates more substrates in human AD brain than in age-matched controls (Tagawa et al., 2015).
For his part, Malinow reported that knocking out PKC-binding protein (PICK1), which interacts with PKCα at the synapse, protected against Aβ synaptic toxicity in mice (Alfonso et al., 2014). Because PKCα also regulates long-term depression (LTD), a key facet of synaptic plasticity, Malinow wondered if the kinase mediated the LTD and synapse loss caused by Aβ.
Co-first author Stephanie Alfonso tested this idea in organotypic hippocampal slices from rats. Bathing the tissue in the general PKC inhibitor bisindolylmaleimide IV prevented synaptic depression brought on by overproduction of Aβ in the slices, but not if PKCα lacked the domain that binds PICK1. Hippocampal slices from PKCα knockout mice were also impervious to Aβ, but if the researchers virally reintroduced PKCα into these cultures, then Aβ once again caused synaptic depression. The results suggested that PKCα, when bound to PICK1, enabled Aβ to weaken synapses.
Is there any evidence that PKCα could have the same effect in people? To answer that, Malinow turned to Tanzi, who oversees the National Institute of Mental Health (NIMH) Alzheimer’s Disease Genetics Initiative, a genomic database of more than 410 families with late-onset AD. From that data set, Tanzi and co-first author Basavaraj Hooli found six families in whom three PKCα mutations in the coding region of the gene largely segregated with LOAD. In four families, seven of nine people with the disease had an M489V mutation; in another family, two of three patients had a V636I mutation; and in one more, an R324W mutation appeared in one of two affected family members. In short, among these families, anyone who carried one of these mutations developed AD. However, in these same families some additional people who carried no PKC mutation also developed AD.
To explore whether these mutations affect PKCα function, co-first author Julia Callender in Newton’s lab tested the activity of all three variants, and of wild-type PKCα in monkey kidney cells. She found that the variants were more active than the wild-type enzyme. Each mutation occurred in or near segments of PKCα that temper activity. M489V lies in a loop near the active site, V636I sits on a C-terminal region that covers the kinase domain (see image above), and R324W occupies the hinge that bridges the kinase domain and the regulatory portion. The researchers concluded that all three mutations potentially tweak the active site to accelerate phosphorylation.
How might enhanced PKCα activity translate to synaptic depression? Newton is unsure, but speculated that when the kinase phosphorylates AMPA receptors, which are known PKC substrates, it triggers their endocytosis. “This could basically exacerbate the Aβ effect,” she said.
Scientists commented to Alzforum that the genetics data falls below statistical significance for association with AD. Since two of the three mutations occur in the general population, and some LOAD family members in this study developed AD without a PKCα variant, it leaves open the possibility that these mutations may not be responsible for disease in these cases.
“This of course does not mean that the data are wrong, but that the paper should stand or fall by the assessment of whether this is truly a convincing candidate gene and not subject to the rules established about achieving genome-wide significance,” said John Hardy, University College London. No AD GWAS has turned up PKCα variants; however, Tanzi noted that these mutations are too rare to do GWAS-type statistics and thus need to be integrated with functional studies. The allele frequencies for M489V and V636I in the 1000 Genomes project are 0.0005 and 0.002, respectively, while R324W is not represented in that data set at all. It is possible that the one or two carriers found in the 1000 Genomes population will develop LOAD later, said Tanzi.
Interestingly, more than 100 PKCα mutations have been linked to cancer, all of them loss-of-function variants that downregulate kinase activity (Antal et al., 2015). “We got excited about our findings because it underscored the critical role of the precise balance in the activity of PKC for normal cellular homeostasis,” said Newton. “Too much activity and you might get a degenerative disease like Alzheimer’s; not enough activity and you get cancer.” Indeed, previous work suggests that people with some forms of cancer are protected from AD and vice versa. “Integrating the biological function of PKC into existing molecular networks of AD and cancer may not only help explain the inverse relationship between these devastating diseases, but may also help reveal an Achilles heel for treating them both,” wrote Vivek Venkataramani, University Medical Center Göttingen, Germany, to Alzforum. However, the loss and gain of PKC function does not fully explain that inverse relationship, Venkataramani added. This is true for many reasons, he said, not least being that PKCα mutations in AD are very rare, suggesting other AD etiologies protect from cancer as well (see full comment below).
Could any of the PKC inhibitors tested over decades in cancer clinical trials be of use in AD? PKC inhibitors were being pursued for a while because researchers realized only recently that PKC also suppresses tumors, meaning it’s probably a bad idea to treat cancer by blocking it, Newton said. If too much PKCα activity is the problem in Alzheimer’s, these drugs could be repurposed for that disease, she proposed. Chronic administration of bryostatin causes downregulation of PKC; this drug is in Phase 2 to test its safety and efficacy in people with moderate to severe AD. Since PKC activity may be upregulated in AD generally—not just in people with mutations—these medicines could benefit many patients, Newton said. However, to avoid inhibiting other PKC subtypes that might increase the risk of cancer, researchers might need to find drugs that specifically target PKCα bound to PICK1.—Gwyneth Dickey Zakaib
References
Paper Citations
- da Cruz e Silva OA, Rebelo S, Vieira SI, Gandy S, da Cruz e Silva EF, Greengard P. Enhanced generation of Alzheimer's amyloid-beta following chronic exposure to phorbol ester correlates with differential effects on alpha and epsilon isozymes of protein kinase C. J Neurochem. 2009 Jan;108(2):319-30. Epub 2008 Dec 2 PubMed.
- Lane RF, Gatson JW, Small SA, Ehrlich ME, Gandy S. Protein kinase C and rho activated coiled coil protein kinase 2 (ROCK2) modulate Alzheimer's APP metabolism and phosphorylation of the Vps10-domain protein, SorL1. Mol Neurodegener. 2010;5:62. PubMed.
- Kim T, Hinton DJ, Choi DS. Protein kinase C-regulated aβ production and clearance. Int J Alzheimers Dis. 2011;2011:857368. PubMed.
- Kinouchi T, Sorimachi H, Maruyama K, Mizuno K, Ohno S, Ishiura S, Suzuki K. Conventional protein kinase C (PKC)-alpha and novel PKC epsilon, but not -delta, increase the secretion of an N-terminal fragment of Alzheimer's disease amyloid precursor protein from PKC cDNA transfected 3Y1 fibroblasts. FEBS Lett. 1995 May 8;364(2):203-6. PubMed.
- Tagawa K, Homma H, Saito A, Fujita K, Chen X, Imoto S, Oka T, Ito H, Motoki K, Yoshida C, Hatsuta H, Murayama S, Iwatsubo T, Miyano S, Okazawa H. Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer's disease brain. Hum Mol Genet. 2015 Jan 15;24(2):540-58. Epub 2014 Sep 17 PubMed.
- Alfonso S, Kessels HW, Banos CC, Chan TR, Lin ET, Kumaravel G, Scannevin RH, Rhodes KJ, Huganir R, Guckian KM, Dunah AW, Malinow R. Synapto-depressive effects of amyloid beta require PICK1. Eur J Neurosci. 2014 Apr;39(7):1225-33. PubMed.
- Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015 Jan 29;160(3):489-502. Epub 2015 Jan 22 PubMed.
External Citations
Further Reading
Papers
- Lucke-Wold BP, Turner RC, Logsdon AF, Simpkins JW, Alkon DL, Smith KE, Chen YW, Tan Z, Huber JD, Rosen CL. Common mechanisms of Alzheimer's disease and ischemic stroke: the role of protein kinase C in the progression of age-related neurodegeneration. J Alzheimers Dis. 2015;43(3):711-24. PubMed.
- Kim T, Hinton DJ, Choi DS. Protein kinase C-regulated aβ production and clearance. Int J Alzheimers Dis. 2011;2011:857368. PubMed.
Primary Papers
- Alfonso SI, Callender JA, Hooli B, Antal CE, Mullin K, Sherman MA, Lesné SE, Leitges M, Newton AC, Tanzi RE, Malinow R. Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer's disease. Sci Signal. 2016 May 10;9(427):ra47. PubMed. Expression of concern.
Annotate
To make an annotation you must Login or Register.
Comments
University Wuerzburg
Although the occurrence of both cancer and Alzheimer’s disease (AD) increase exponentially as people age, recent epidemiological studies focused on the curious observation that cancer patients rarely develop AD, and vice versa (Driver et al., 2012). Different studies demonstrated that this relationship may be AD-specific, as no significant correlation could be seen to non-AD diseases, such as vascular dementia (Roe et al., 2010; Musicco et al., 2013). This inverse correlation implicates the possibility that both diseases might be regulated in opposite directions at the molecular level, and that a deeper understanding may allow the development of new treatment strategies for both diseases.
The recent study from the Malinow lab highlights the involvement of protein kinase Cα (PKCα) in AD pathology (Alfonso et al., 2016). Previously, the group reported that the PKC-binding protein (PICK1) was required for Aβ-induced synaptic depression (Alfonso et al., 2014). In the current study they now revealed the direct and selective contribution of overactivated PKCα in Aβ-mediated synaptotoxicity by using pharmacological inhibition and ex vivo analysis of PKCα-knockout mice. Intriguingly, analyzing whole-sequencing data sets from Rudi Tanzi’s Alzheimer’s Disease Genetics Initiative Study identified three unique PKCα mutations that significantly co-segregate with AD development in families with the disease. In line with their hypothesis that increased PKCα activity mediates the synaptotoxic effects of Aβ, functional cell-based analysis revealed that all three AD-associated PKCα variants were gain-of-function mutations displaying increased kinase activity.
What about PKCα in cancer? PKC was initially described as receptor of the tumor-promoting phorbol ester, strengthening the dogma that activation of PKC drives tumorigenesis (Castagna et al., 1982). However, the group of Alexandra Newton (who was also involved in the Malinow study) challenged this dogma and revealed that most of the cancer-associated mutations of the PKC family (including the conventional α-isoform) have lost their function (Antal et al., 2015). Detailed characterization of 46 of these PKC mutations showed that the majority of these variants resulted in suppressed/abolished PKC activity. Moreover, genetic restoration of heterozygote loss-of-PKC-function nicely resulted in a reduced tumor volume, suggesting a tumor-suppressive function of PKC. This study may also explain why several PKC inhibitors failed in clinical trials. Notably, in the regard of over-activated PKC function in AD, these inhibitors may serve as a therapeutic option, at least for the subset of patients with mutated gain-of-function PKCα.
The question now arises whether the loss/gain-of-function of PKCα explains the inverse correlation between AD and cancer. It is very intriguing that in AD only PKCα variants with gain of function were identified, whereas in the plethora of analysed cancers all PKC mutations had a loss of function or at least no gain of function. However, these opposing functions of PKC cannot solely explain the inverse AD/cancer-relationship. PKC is frequently mutated in human cancer, whereas all three identified PKC variants in AD patients were rare. Moreover, the majority of identified PKC mutations are heterozygous with an allele frequency varying from 0.05-0.67, indicating that these kinds of mutations are acquired in the later steps of tumorigenesis. In contrast, overactivated PKCα is directly contributing to Aβ-mediated synaptotoxicity, which is considered an early event in AD pathogenesis. More studies are needed to decipher how PKC functions as a tumor-suppressor and its possible connections with p53 tumor-suppressor and Hippo pathway regulators. Furthermore, recent studies also demonstrated that Pin1(Driver et al., 2015; Pastorino et al., 2006) and the amyloid precursor protein (APP) also present critical but opposite roles in AD pathogenesis and cancer. In the context of APP, we and others have demonstrated that APP and its α-cleaved sAPPα fragment are not only overexpressed in a plethora of solid and hematological cancers, but also are highly relevant for cancer survival and tumorigenesis (Venkataramani et al., 2010; Venkataramani et al., 2012). Integrating the biological function of PKC into existing molecular networks of AD and cancer may not only help explain the inverse relationship between these devastating diseases, but may also help reveal an Achilles heel for treating them both.
References:
Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T, Kiel DP, Lu KP, Seshadri S, Wolf PA. Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. BMJ. 2012 Mar 12;344:e1442. PubMed.
Roe CM, Fitzpatrick AL, Xiong C, Sieh W, Kuller L, Miller JP, Williams MM, Kopan R, Behrens MI, Morris JC. Cancer linked to Alzheimer disease but not vascular dementia. Neurology. 2010 Jan 12;74(2):106-12. Epub 2009 Dec 23 PubMed.
Musicco M, Adorni F, Di Santo S, Prinelli F, Pettenati C, Caltagirone C, Palmer K, Russo A. Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology. 2013 Jul 23;81(4):322-8. Epub 2013 Jul 10 PubMed.
Alfonso SI, Callender JA, Hooli B, Antal CE, Mullin K, Sherman MA, Lesné SE, Leitges M, Newton AC, Tanzi RE, Malinow R. Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer's disease. Sci Signal. 2016 May 10;9(427):ra47. PubMed. Expression of concern.
Alfonso S, Kessels HW, Banos CC, Chan TR, Lin ET, Kumaravel G, Scannevin RH, Rhodes KJ, Huganir R, Guckian KM, Dunah AW, Malinow R. Synapto-depressive effects of amyloid beta require PICK1. Eur J Neurosci. 2014 Apr;39(7):1225-33. PubMed.
Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982 Jul 10;257(13):7847-51. PubMed.
Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase's role as tumor suppressor. Cell. 2015 Jan 29;160(3):489-502. Epub 2015 Jan 22 PubMed.
Driver JA, Zhou XZ, Lu KP. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer's disease. Biochim Biophys Acta. 2015 Oct;1850(10):2069-76. Epub 2015 Jan 10 PubMed.
Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006 Mar 23;440(7083):528-34. PubMed.
Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, Wirths O, Bayer TA. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J Biol Chem. 2010 Apr 2;285(14):10678-89. Epub 2010 Feb 9 PubMed.
Venkataramani V, Thiele K, Behnes CL, Wulf GG, Thelen P, Opitz L, Salinas-Riester G, Wirths O, Bayer TA, Schweyer S. Amyloid precursor protein is a biomarker for transformed human pluripotent stem cells. Am J Pathol. 2012 Apr;180(4):1636-52. Epub 2012 Feb 1 PubMed.
Icahn School of Medicine at Mount Sinai
In 1990, a series of independent papers converged to implicate a role for protein kinase C (PKC) in AD. Eliezer Masliah and Greg Cole, working in Tsunao Saitoh’s lab, discovered deficiencies in PKC activity in AD brain and fibroblasts (Masliah et al., 1990; Masliah et al., 1991). Working with Paul Greengard, I had discovered the direct phosphorylation of APP by PKC in 1988 (Gandy et al., 1988); then, Joseph Buxbaum worked together with us and we extended this observation and showed that PKC activation stimulates activity of the α-secretase, increasing sAPPα release (Buxbaum et al., 1990) and reducing Aβ generation (Buxbaum et al., 1992). This work was instantly confirmed in identical studies that emerged from the collaborative program involving Roger Nitsch, John Growdon, Dick Wurtman, Christian Haass, and Dennis Selkoe (Nitsch et al., 1992; Hung et al., 1993). Conveniently, Du-Sup Choi and Robert Messing demonstrated that PKC activates a second Aβ-reducing pathway by stimulating degradation by endothelin converting enzyme (ECE) (Choi et al., 2006). Taken together, all these data accumulating over two decades pointed to PKC activation as a potential therapeutic strategy for AD, and, indeed, forms part of the basis of Dan Alkon’s clinical trial with the PKC activator bryostatin (Etcheberrigaray et al., 2004).
This has been by no means a straightforward path because: (1) PKC levels rapidly downregulate following stimulation; (2) the main laboratory activators of PKC (or inactivators of the corresponding protein phosphatase) are tumor promoters; and (3) the promoter of APP is sensitive to PKC activation. Odete da Cruz e Silva, working with the late Edgar da Cruz e Silva, in a collaboration between the Greengard lab and my lab, showed that, somewhat unexpectedly, with chronic PKC activation, Aβ accumulates (implying that the APP transcriptional activation outstrips both the differential processing by α-secretase and the activated Aβ degradation by ECE) and PKC levels are downregulated (da Cruz e Silva, 2009). This new discovery by Rudy Tanzi, Roberto Malinow, and Alexandra Newton of mutations in PKC that cause gain of function would potentially fall into this latter category, doing more harm than good.
Moreover, this new synaptic role for PKC in modulating Aβ-induced synaptic depression potentially yields a genetic double whammy if the overactive PKC clinical mutation drives both Aβ accumulation and Aβ-induced synaptic depression. This discovery provides yet another illustration of how the initiation of the pathway to AD might begin at one or multiple levels culminating in a concatenation of pathology involving amyloidosis, immune dysfunction, tauopathy, and failure and death of synapses and neurons.
References:
Masliah E, Cole G, Shimohama S, Hansen L, DeTeresa R, Terry RD, Saitoh T. Differential involvement of protein kinase C isozymes in Alzheimer's disease. J Neurosci. 1990 Jul;10(7):2113-24. PubMed.
Masliah E, Cole GM, Hansen LA, Mallory M, Albright T, Terry RD, Saitoh T. Protein kinase C alteration is an early biochemical marker in Alzheimer's disease. J Neurosci. 1991 Sep;11(9):2759-67. PubMed.
Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci U S A. 1988 Aug;85(16):6218-21. PubMed.
Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990 Aug;87(15):6003-6. PubMed.
Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci U S A. 1992 Nov 1;89(21):10075-8. PubMed.
Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992 Oct 9;258(5080):304-7. PubMed.
Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem. 1993 Nov 5;268(31):22959-62. PubMed.
Choi DS, Wang D, Yu GQ, Zhu G, Kharazia VN, Paredes JP, Chang WS, Deitchman JK, Mucke L, Messing RO. PKCepsilon increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc Natl Acad Sci U S A. 2006 May 23;103(21):8215-20. Epub 2006 May 12 PubMed.
Etcheberrigaray R, Tan M, Dewachter I, Kuipéri C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Therapeutic effects of PKC activators in Alzheimer's disease transgenic mice. Proc Natl Acad Sci U S A. 2004 Jul 27;101(30):11141-6. Epub 2004 Jul 19 PubMed.
da Cruz e Silva OA, Rebelo S, Vieira SI, Gandy S, da Cruz e Silva EF, Greengard P. Enhanced generation of Alzheimer's amyloid-beta following chronic exposure to phorbol ester correlates with differential effects on alpha and epsilon isozymes of protein kinase C. J Neurochem. 2009 Jan;108(2):319-30. Epub 2008 Dec 2 PubMed.
Make a Comment
To make a comment you must login or register.